赵方庆 博士/研究员(中国科学院北京生命科学研究院)
报告主题:环形非编码RNA计算方法学研究

嘉宾简介:
中国科学院“百人计划”入选者、国家自然科学基金优青基金和北京市杰出青年基金获得者。现为中科院北京生科究院科研部副主任、中国生物工程学会计算生物学与生物信息学专业委员会副主任,Briefings in Bioinformatics, Hereditas, BMC Evolutionary Biology, Genomics, Proteomics & Bioinformatics等国际刊物编委。近年来,在Nature Communications, Gut, Genome Biology, Trends in Genetics, ISME J, Current Biology和Nucleic Acids Res 等刊物上发表通讯作者论文30余篇,平均影响因子超过10,其中多篇入选ESI高被引论文;两次荣获“中国科学院优秀导师奖”(2017,2018);培养的研究生已有4人次获得“中科院院长奖”和“中科院优秀博士学位论文”。
代表性成果介绍:
Cell Reports:多物种circRNA表达数据库circAtlas
2019年3月19日,Cell Reports杂志在线发表了中科院北京生命科学研究院赵方庆研究员团队的一项工作,鉴定了人类及其他多种动物正常组织的转录组,组装了其中大部分circRNA的全长序列,汇总形成circRNA数据库(circAtlas)。
circAtlas数据库目前已收录了包括人(Homo sapiens),猕猴(Macaca mulatta),小鼠(Mus musculus),褐家鼠(Rattus norvegicus),野猪(Sus scrofa)和鸡(Gallus gallus)。可变剪切分析表明在人,猕猴和小鼠中四种可变剪切均能检测到。很多情况下同一个基因存在多种circRNA产物,但作者从所获得的的表达数据中统计发现大部分基因都有一种主要存在的circRNA分子。表达相关性分析表明circRNA的表达量与RNA结合蛋白相关性非常强,这也可能是造成circRNA组织特异性表达的一个原因。比较不同物种中circRNA的表达特征后发现在不同物种中存在一类重叠的直系同源circRNA分子(overlapped orthologs,OO-type),就是指在不同物种中剪切位置高度保守的circRNA分子。直系同源的mRNA表达情况在物种间的一致性明显比OO-type circRNA高。有少数的OO-type circRNA表达丰度甚至远高于来源基因,暗示这些分子或许具有特殊的重要功能。(参考文献[1])
推荐阅读:circAtlas: 多物种circRNA表达数据库

Genome Medicine:全长circRNA捕获分析方法
2019年1月19日,Genome Medicine杂志发表了中科院北京生命科学研究院赵方庆研究员团队的一项工作,介绍一种分析全长circRNA的技术方法。
本文中提出了识别环状RNA全长序列的方法CIRI-full,相比基于BSJ的方法,RO方法对低表达丰度的环状RNA 更敏感。 利用FSG的方法精确识别和量化环状RNA的转录本(isoform)。本文分析了 6个物种(包括人,恒河猴,小鼠,大鼠,兔,鸡)大脑全长环状RNA。相比BSJ,CIRI-full获得的转录本(isoform)能过滤假阳性的差异表达环状RNA。(参考文献[2])
推荐阅读:全长环状RNA捕获与定量
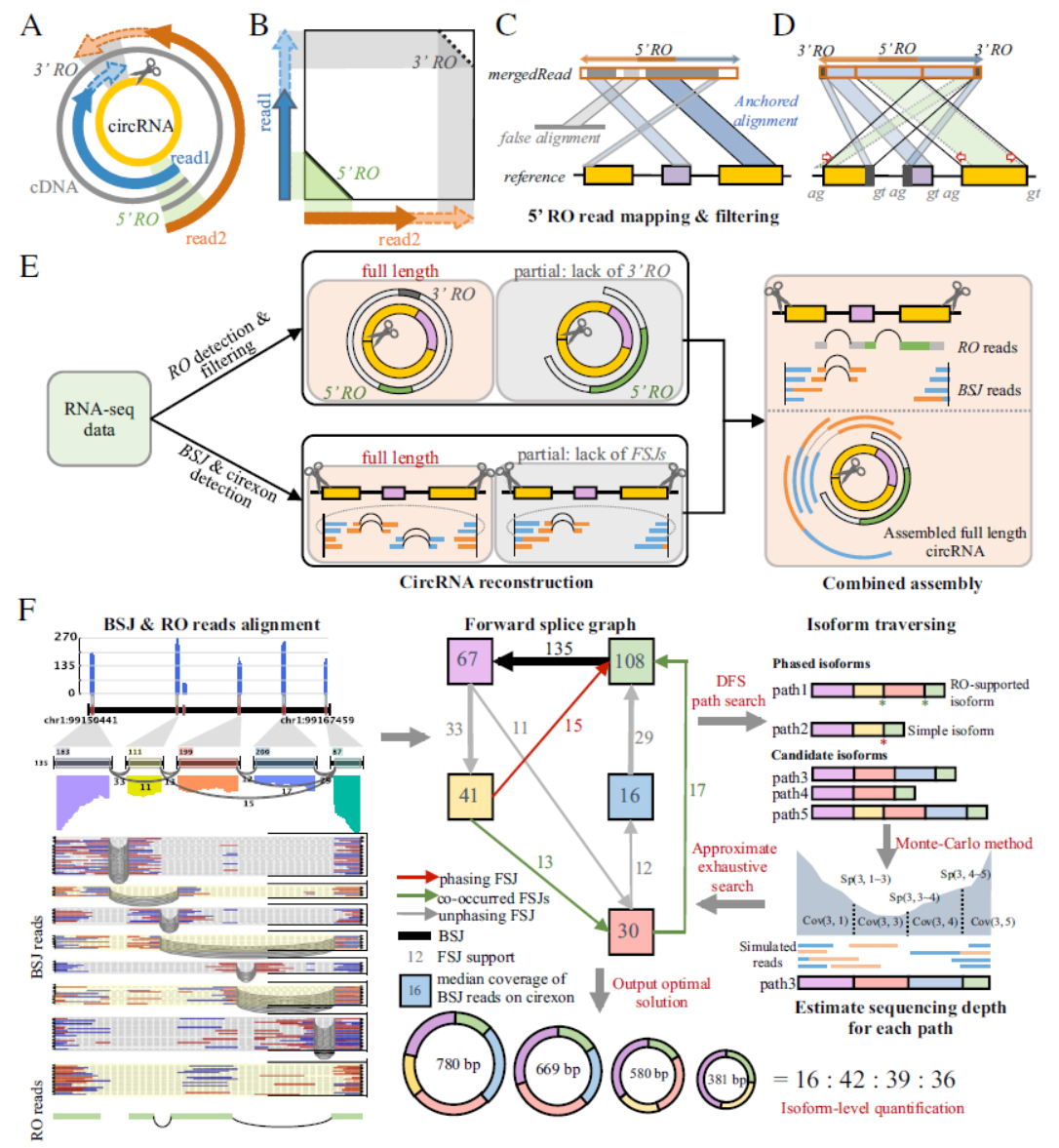
RO检测全长circRNA技术流程 (参考文献[2])
Briefings in Bioinformatics:基于多种子匹配的circRNA鉴定方法
2018年9月28日,Briefings in Bioinformatics发表了中科院北京生命科学研究院赵方庆研究员团队的一篇文章,报道一种基于多种子序列匹配的circRNA鉴定方法。
circRNA测序数据的分析是研究circRNA的关键,但此前用于分析circRNA的工具都或多或少存在一些缺陷。本文中作者提出了一种改进的多线程检测工具CIRI2,它使用基于多种子匹配的自适应最大似然估计来识别反向拼接结读数并过滤从重复序列和映射误差导出的误报。根据RNase R处理样品的实际数据建立了客观评估标准,并系统地比较了10种circRNA检测工具,证明CIRI2优于其先前版本CIRI和所有其他广泛使用的工具,具有非常均衡的灵敏度,可靠性,持续时间和RAM 用法。(参考文献[10])
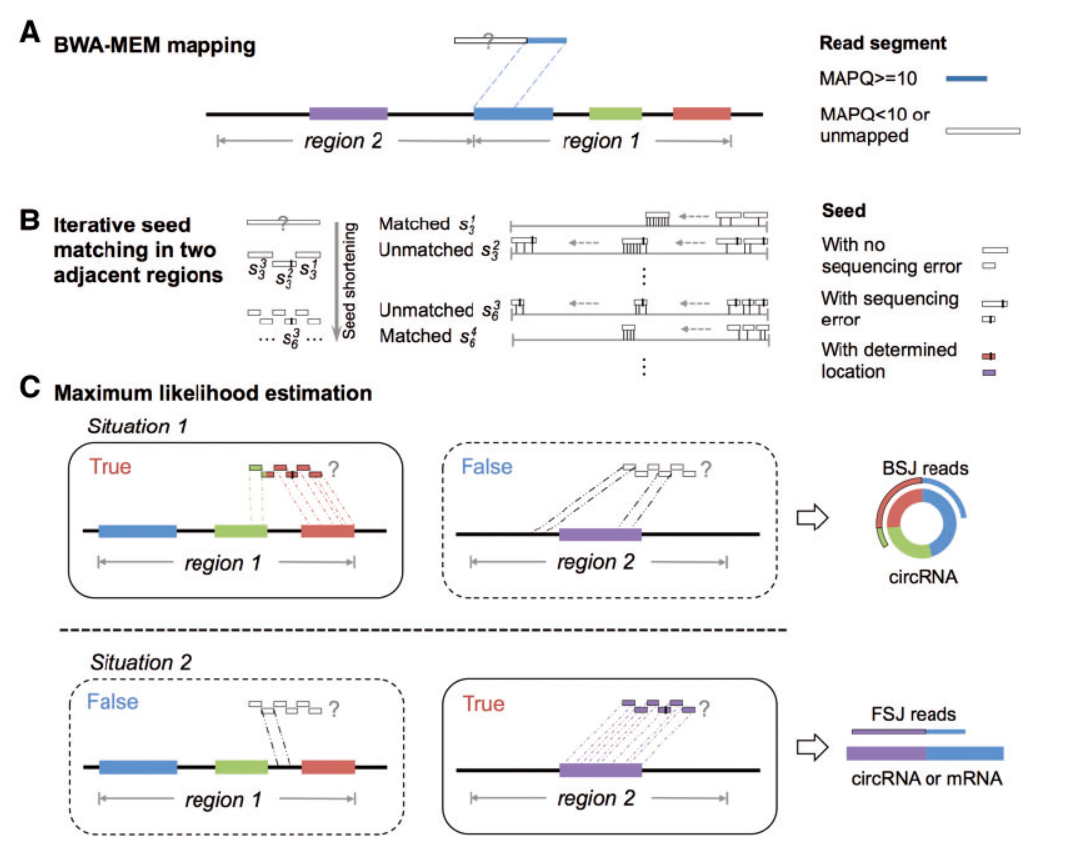
CIRI2算法流程示意图 (参考文献[10])
Nature Communications:circRNA可变剪切的发现与分析
2016年6月28日,中国科学院北京生命科学研究院计算基因组实验室赵方庆教授团队在国际知名杂志Nature Communication发表论文,利用环状RNA预测分析结合长片段测序技术,首次实现了针对环状RNA可变剪切状态进行了系统研究。
环状RNA本身就是可变剪切的产物,但每个环状RNA分子内部是否仍有可变剪切方式?环状RNA的二级结构如何?这些都是非常有趣的问题。赵方庆教授团队基于该问题,设计了一种新的整合环状RNA结构与序列信息的技术分析流程,作者称之为CIRI-AS。基于此技术,作者在10种人类细胞以及62个果蝇不同组织来源的样品中分析了环状RNA可变剪切的情况,结果表明理论上的四大类可变剪切方式在环状RNA中均普遍存在。这一发现大大增进了对环状RNA的认识,也为环状RNA研究提供了新思路和新工具。(参考文献[12])
推荐阅读:从RNA结构预测角度探索环状RNA功能新进展
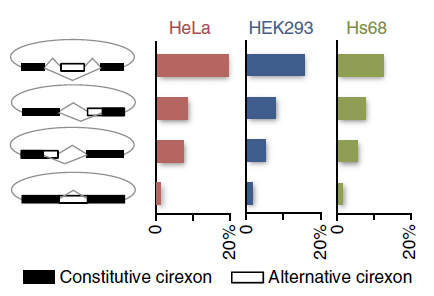
环状RNA中存在四种可变剪切方式,但比例相差很大 (参考文献[12])
Genome Biology:CIRI算法工具
2015年1月13日,Genome Research杂志发表了中国科学院北京生命科学研究院计算基因组实验室赵方庆教授团队开发的circRNA分析工具:CIRI。CIRI是常用的circRNA测序数据比对分析的工具,得到了同行的广泛应用。(参考文献[15])

赵方庆教授长期致力于circRNA算法工具和数据分析的技术开发工作,他们开发的CIRI,CIRI2工具得到了同行的广泛应用和高度认可。他们也是最早发现和报道circRNA可变剪切的团队之一。他们开发的RO技术进行全长circRNA的鉴定分析为circRNA可变剪切和全长序列的鉴定提供了有效的技术体系。circAtlas数据库整合了大量高质量的数据资源,为同行开展cricRNA研究提供了非常方便有效的在线分析工具和数据资源。
代表性研究成果列表
1. Ji P, Wu W, Chen S, Zheng Y, Zhou L, Zhang J, Cheng H, Yan J, Zhang S, Yang P, Zhao F*. Expanded Expression Landscape and Prioritization of Circular RNAs in Mammals. Cell Reports. 2019, 26(12):3444-3460
2. Zheng Y, Ji P, Chen S, Hou L & Zhao F*. Reconstruction of full-length circular RNAs enables isoform-level quantification. Genome Medicine, 2019 Jan 19;11(1):2
3. He M, Wang J, Fan X, Liu X, Shi W, Huang N, Zhao F* & Miao M*. Genetic basis for the establishment of endosymbiosis in Paramecium. ISME J. 2019,13(5):1360-1369
4. Wang J, Zheng J, Shi W, Du N, Xu X, Zhang Y, Ji P, Zhang F, Jia Z, Wang Y, Zheng Z, Zhang H & Zhao F*. Dysbiosis of maternal and neonatal microbiota associated with gestational diabetes mellitus. Gut, 2018, 67:1614-1625.
5. Gao Y & Zhao F*. Computational strategies for exploring circular RNAs. Trends in Genetics, 2018, 34(5): 389-400.
6. Zhou L & Zhao F*. Prioritization and functional assessment of noncoding variants associated with complex diseases. Genome Medicine 2018, 10:53.
7. Teng H, Zhang Y, Shi C, Mao F, Cai W, Lu L, Zhao F*, Sun Z* & Zhang J*. Population genomics reveals speciation and introgression between Brown Norway rats and their sibling species. Molecular Biology Evolution 2017, 34(9):2214-2228.
8. Ji P, Zhang Y, Wang J & Zhao F*. MetaSort untangles metagneome assembly by reducing microbial community complexity. Nature Communications, 2017, 8:14306.
9. Shi W, Ji P & Zhao F*. The combination of direct and paired link graphs boosts repetitive genome assembly. Nucleic Acids Res 2017, 45 (6): e43.
10. Gao Y, Zhang J & Zhao F*. Circular RNA identification based on multiple seed matching. Briefings in Bioinformatics 2017, DOI: 10.1093/bib/bbx014.
11. Peng G, Ji P & Zhao F*. A novel codon-based de Bruijn graph algorithm for gene construction from unassembled transcriptomes. Genome Biology 2016, 17:232.
12. Gao Y, Wang J, Zheng Y, Zhang J, Chen S & Zhao F*. Comprehensive identification of internal structure and alternative splicing events in circular RNAs. Nature Communications 2016, 7:12060.
13. Zhang Y, Ji P, Wang J & Zhao F*. RiboFR-Seq: a novel approach to linking 16S rRNA amplicon profiles to metagenomes. Nucleic Acids Res. 2016, 44:e99.
14. Zhang Z, Xu D, Wang L, Hao J, Wang J, Zhou X, Wang W, Qiu Q, Huang X, Zhou J, Long R*, Zhao F* & Shi P*. Convergent evolution of rumen microbiomes in high-altitude mammals. Current Biology, 2016, 26(14): 1873-1879.
15. Gao Y, Wang J & Zhao F*. CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biology. 2015, 16:4.
16. Zhao H & Zhao F*. BreakSeek: a breakpoint-based algorithm for full spectral range INDEL detection. Nucleic Acids Res. 2015, 43 (14):6701-6703.
17. Ye N^*, Zhang X^, Miao M^, Fan X^, Zheng Y, Xu D, Wang J, Zhou L, Wang D, Gao Y, Wang Y, Shi W, Ji P, Li D, Guan Z, Shao C, Zhuang Z, Gao Z, Qi J* & Zhao F*. Saccharina genomes provide novel insight into kelp biology. Nature Communications. 2015, 6: 6986.
18. Wang J, Gao Y & Zhao F*. Phage-bacteria interaction network in human oral microbiome. Environmental Microbiology. 2016, 18(7):2143-2158.