引言
众所周知,大量物种全基因组范围内发现了许多由外显子反向剪接产生的环状RNA (circRNA)。尽管大部分都是一些表达水平低的circRNA,但一些表达相对丰富的circRNA可以在各种生物过程中发挥调节作用。
与线性RNA的共线性顺序剪接相比,环状RNA中具有非共线性的反向剪接(BSJ)特征。环状RNA在很大程度上与同源线性RNA共表达,且序列几乎完全重叠,因此在全基因组范围内精确注释和量化环状RNA一直具有挑战性。
为此,复旦大学生物医学研究院特聘研究员杨力在Trends in Genetics (IF=11.821) 期刊上发表了一篇题为”Approaches and challenges in genome-wide circular RNA identification and quantification”的综述,讨论了circRNA不同的纯化和富集策略、不同的测序方法以及全基因组范围内circRNA分析相应的计算方法,并强调了当使用不同的策略时,circRNA的准确定量对于跨样本比较的重要性。
主要内容
1、circRNA分析的不同富集方法和测序平台
早期的全转录组学研究方法是通过oligo(dT)珠富集的聚腺苷化[poly(A)+] RNA实现的(称为poly(A)+ RNA-seq) (图1B),但无法用来大规模识别环状转录本,因此只能通过其他环状RNA富集策略来实现。一方面可以通过收集与oligo(dT)无关的片段并进一步消耗冗余RNA,提取非聚腺苷化[poly(A)-]的RNA进行深度测序 (称为poly(A)- RNA-seq) (图1B)。另一方面,对同时含有poly(A)+和poly(A)-的转录本进行rRNA消除后再进行深度测序,称为ribo-RNA-seq (图1B)。这两种手段均可用于分析circRNA和内含子环状RNA(ciRNA),并且能检测到大约10倍以上的BSJ reads (图1D),另外还可用于环状RNA与其同源线性RNA的比较。此外,进行RNase R处理后,circRNA和ciRNA可在深度测序中被显著富集和沉默,称为RNaseR RNA-seq (图1B)。最近几年兴起的Oxford Nanopore等长读长测序平台也已经实现了对circRNA的识别分析 (图1B)。
在RNase R处理的短读长和长读长测序数据集中,可以检测到的BSJ reads数比非RNase R处理的多3到10倍(图1D)。由于RNase R处理消除了反式剪接和逆转录酶模板切换导致的假阳性,因此它已被广泛用于circRNA的验证。
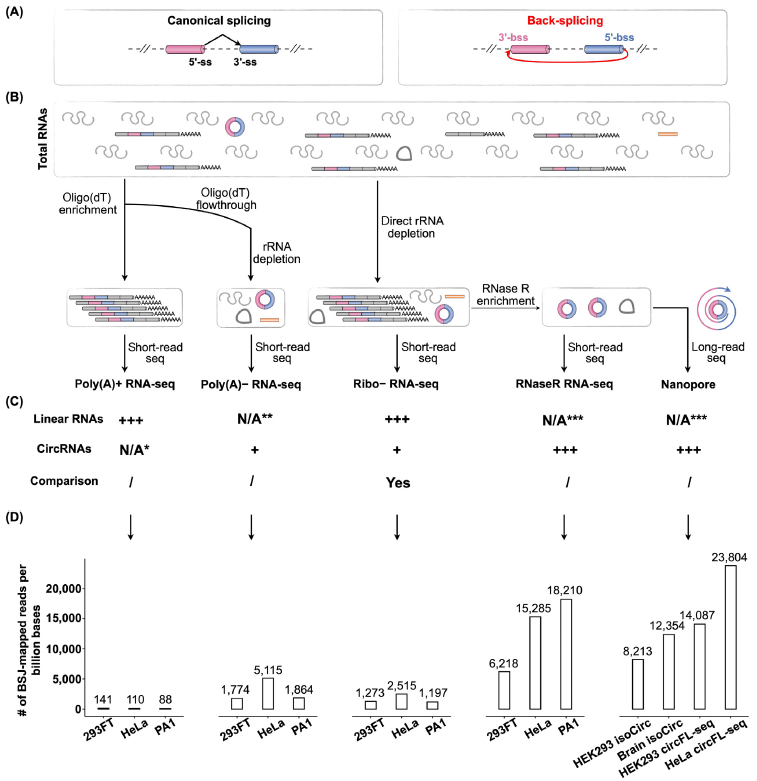 图1 circRNA分析的不同富集方法和测序平台
图1 circRNA分析的不同富集方法和测序平台
2、circRNA识别分析方法
几乎所有关于circRNA可靠注释的计算管道都采用统一的原则:识别比对到BSJ的测序读段。到目前为止,已经开发了十几种计算方法,用于从短读长RNA-seq数据集中分析circRNA,而根据如何识别比对到BSJ的reads,这些计算方法可以简单地分为两类:基于融合读段 (fusion reads, 图2A) 和基于伪参考基因组 (pseudo reference, 图2B) 的方法。
基于融合读段的方法直接将测序读段序列与基因组参考序列比对,然后以非线性方式检测融合(或嵌合)读段序列,常用的代表性工具包括CIRCexplorer3/CLEAR、DCC和MapSplice等。此外,一些软件还开发了独特的策略来重新发现含有未注释外显子的环状RNA,比如CIRCexplorer3使用StringTie或Cufflinks等工具来组装新的转录本,而CIRI2和find_circ则使用剪接信号、剪接位点距离和其他一些因素来实现类似的目标 (图2C)。
基于伪参考基因组的方法需要在比对前根据已有的基因注释数据初步构建伪BSJ参考。比如KNIFE和NCLScan等软件,用于识别比对到伪BSJ参考的reads (图2C)。为了消除潜在的假阳性,这些管道通常首先将reads与基因组和转录组参考注释匹配,然后再与伪BSJ参考注释匹配 (图2B)。由于它们依赖于已知的基因注释,基于伪参考的方法可能不适合circRNA的从头发现。
也有人提出,使用多种工具检测circRNA可以提供更准确的结果,从而出现了合并各种工具的新软件。例如,CircComPara2集成了CIRCexplorer2、find_circ和CIRI,而CircRNAwrap则集成了多达8个circRNA检测工具和多个circRNA分析工具。
除了短读长测序,目前也已经报道了四种类型的长读长测序数据分析工具 (图2D)。其中,CIRI-long、cirFL-seq、isoCirc需要滚动环反转录(RCRT)或滚动环扩增(RCA)过程,然后识别循环一致序列(CCS)或一致序列。而circNick-LRS跳过了RCRT或RCA步骤,直接对circRNA进行长读长测序。与短读长测序相比,长读长测序在检测全长circRNA和识别同分异构体方面具有明显优势,缺点则是成本高和错误率高。
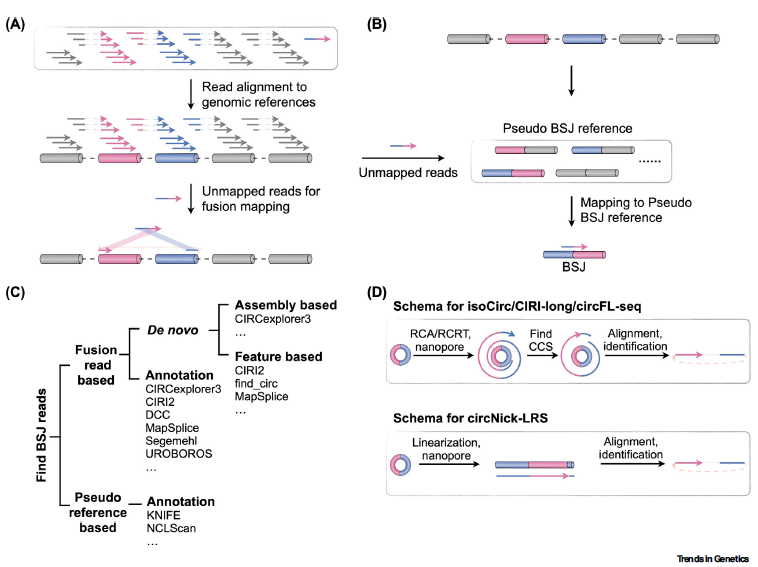 图2 调用BSJ 比对reads进行circRNA注释
图2 调用BSJ 比对reads进行circRNA注释
3、circRNA定量和跨样本比较的挑战
根据circRNA BSJ reads识别比对的统一原则,BSJ比对reads的原始数量自然被用于量化circRNA。然而,由于不同的数据集和研究通常采用不同的测序深度,因此使用BSJ比对reads的绝对数量进行跨样本比较可能存在偏差。例如,随着从同一HLF ribo- RNA-seq数据集中随机提取的20M至150M reads的测序深度的增加,高表达circRNA数量也相应增加 (图3A,左)。而随着测序深度的增加,可以识别出更多表达水平较低的circRNA (图3A,右)。
此外,不同的纯化和富集策略也会影响测出的BSJ比对reads的数量,比如经过或未经过RNase R处理的数据集,以及不同的RNase R处理条件下的数据集量化circRNA及其跨样本的比较可能存在偏差。
最后,同样重要的一点是,在评估circRNA表达时应考虑线性RNA的表达,因为circRNA及其同源线性RNA共存于同一基因组位点。环状RNA的功能研究可能会被高度表达的线性RNA同源异构体所掩盖,因为它们的序列几乎完全重叠。此外,直接比较环状RNA与同源线性RNA的表达是困难的,这不仅是因为它们的序列相似,还因为用于定量环状RNA和线性RNA的策略不同。为了解决这一问题,已有研究报道了分别使用CIRCexplorer3/CLEAR、CIRI2、CIRIquant和DCC软件对BSJ比对reads和与共线性外显子-外显子连接处比对reads进行量化,以评估环状或线性RNA的表达水平(图3C)。以同源线性RNA的表达为背景,可以选择高表达的环状RNA和低表达的线性RNA进行后续的功能研究。
然而,除了一小部分外,绝大多数circRNA的丰度远低于其同源线性RNA,并且已经证明了使用高表达线性RNA来确保表征功能性circRNA的有用方法。
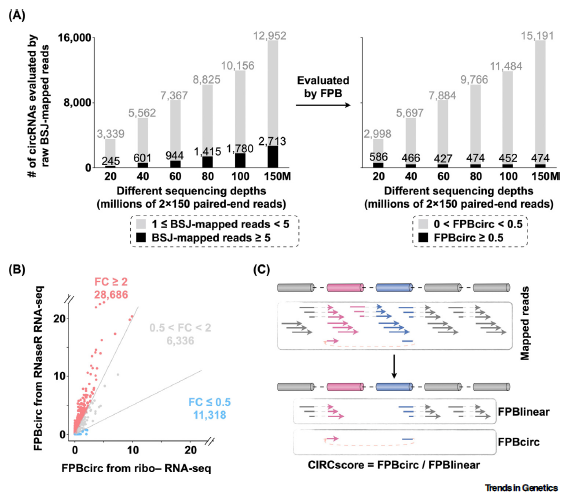 图3 环状RNA的全基因组定量
图3 环状RNA的全基因组定量
总结与展望
在过去的十年中,已有多种(环状)RNA富集策略和测序技术用于实现circRNA的全基因组表征。但全基因组范围内对circRNA的精确识别仍具有挑战性。此外,如何在单细胞水平上有效和精确地分析环状RNA也是一个难题。
全基因组环状RNA分析及其精确定量的复杂性源于环状和线性RNA转录物与重叠序列的共表达。不同的转录组富集策略、测序技术和计算方法促进了circRNA的识别和跨样本比较,以检查它们在各种生物环境中的功能作用。因此,进一步开发专为环状RNA设计的创新技术,将会让我们对这些具有独特环状结构的迷人非编码RNA分子有更深入的理解,以及评估它们在诊断和治疗中的潜在应用。
参考文献
Ma XK, Zhai SN, Yang L. Approaches and challenges in genome-wide circular RNA identification and quantification. Trends Genet. 2023