近日,Nature子刊 Cell Death & Differentiation先后在线发表了两篇circRNA相关研究论文:

本篇文章的通讯作者是青岛大学转化医学院发育心脏病学中心主任王昆教授,介绍发现心肌缺血再灌注模型中一种自噬相关circRNA(ACR)能够通过结合Dnmt3b抑制Pink1基因的甲基化,促进Pink1的表达,Pink1可促进FAM65B的磷酸化并抑制自噬的进程,在心肌缺血再灌注损伤过程中保护细胞[1]。

本篇文章的通讯作者是华中科技大学同济医学院附属协和医院童强松和郑丽端。本文介绍AGO2基因内含子来源的circRNA(circAGO2)可结合HuR并促进其在靶基因3’UTR富集,阻止AGO2复合物结合,促进了靶基因的稳定性,促进肿瘤进程[2]。
下面让我们分别学习一下两篇文章的故事吧:
ACR 参与自噬调控
自噬是重要的细胞生理活动过程,circRNA与自噬的关系之前已有相关报道,包括东南大学姚红红教授分别于2017年8月份和12月份发表于Autophagy杂志的两篇文章,分别介绍了circHIPK2/miR-124-2HG/SIGMAR1轴通过调控自噬和ER Stress通路调控星形胶质细胞活化作用[3],circRNA HECW2/miR-30D调控ATG5和Notch1通路[4]。在本篇文章中,作者通过芯片法分析了心肌缺血再灌注(I/R)后小鼠心肌circRNA表达差异,发现mmu_circRNA_006636在I/R后显著下调,作者将其命名为自噬相关circRNA(Autophagy related circular RNA, ACR)
进一步,作者通过RNase R处理前后QPCR证明ACR的环形结构,过表达ACR等实验证明ACR过表达可抑制细胞自噬过程。
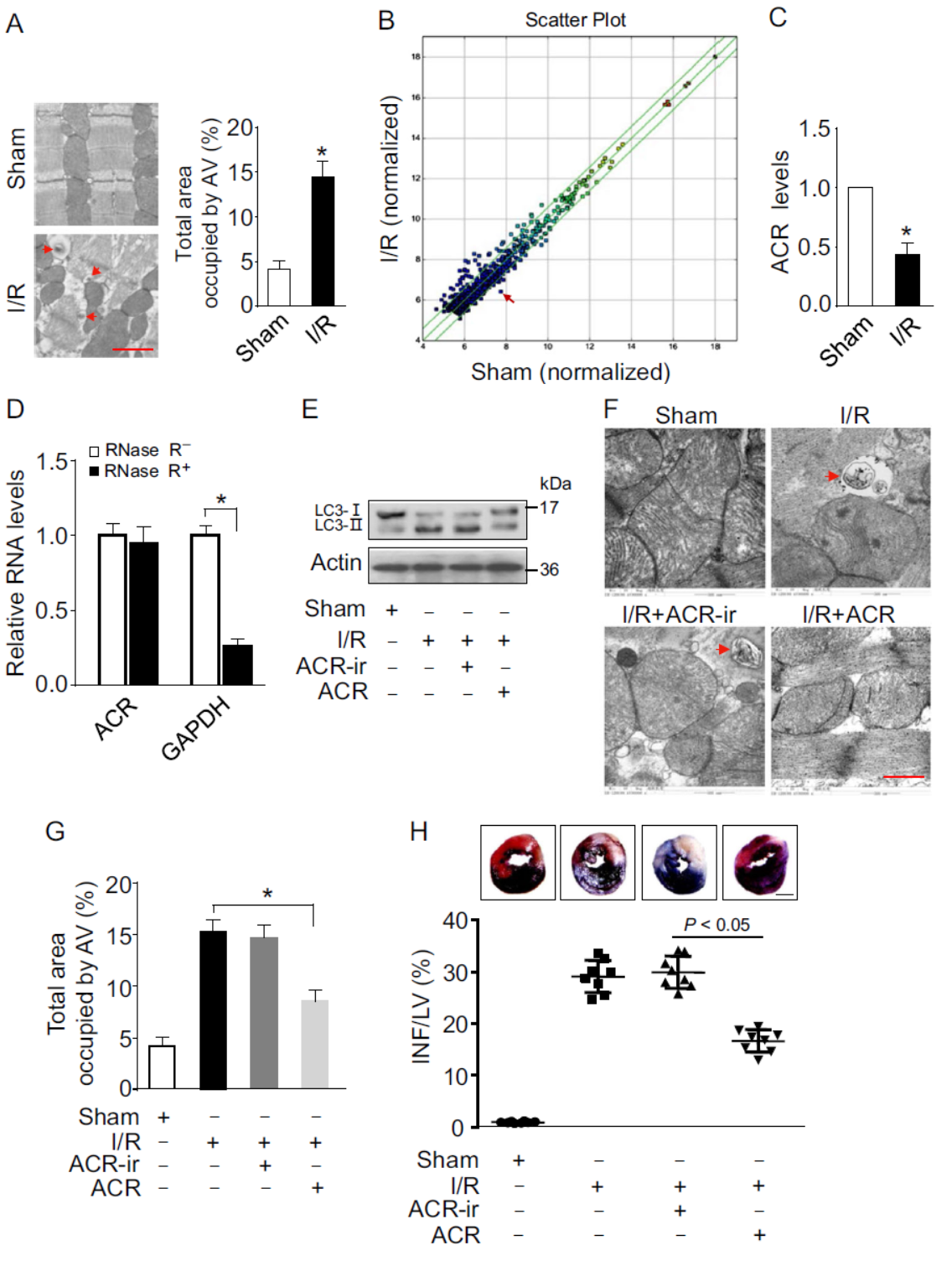
图1 ACR与细胞自噬有关 (引自[1])
缺氧/复氧(A/R)处理后ACR的表达会显著下降,过表达ACR后A/R处理导致的LC3聚集斑点有所减少,LC3-II也比对照组有所下降,说明过表达ACR可抑制自噬过程。电子显微镜分析后也表明过表达ACR后由A/R处理诱导的自噬小泡数量减少。
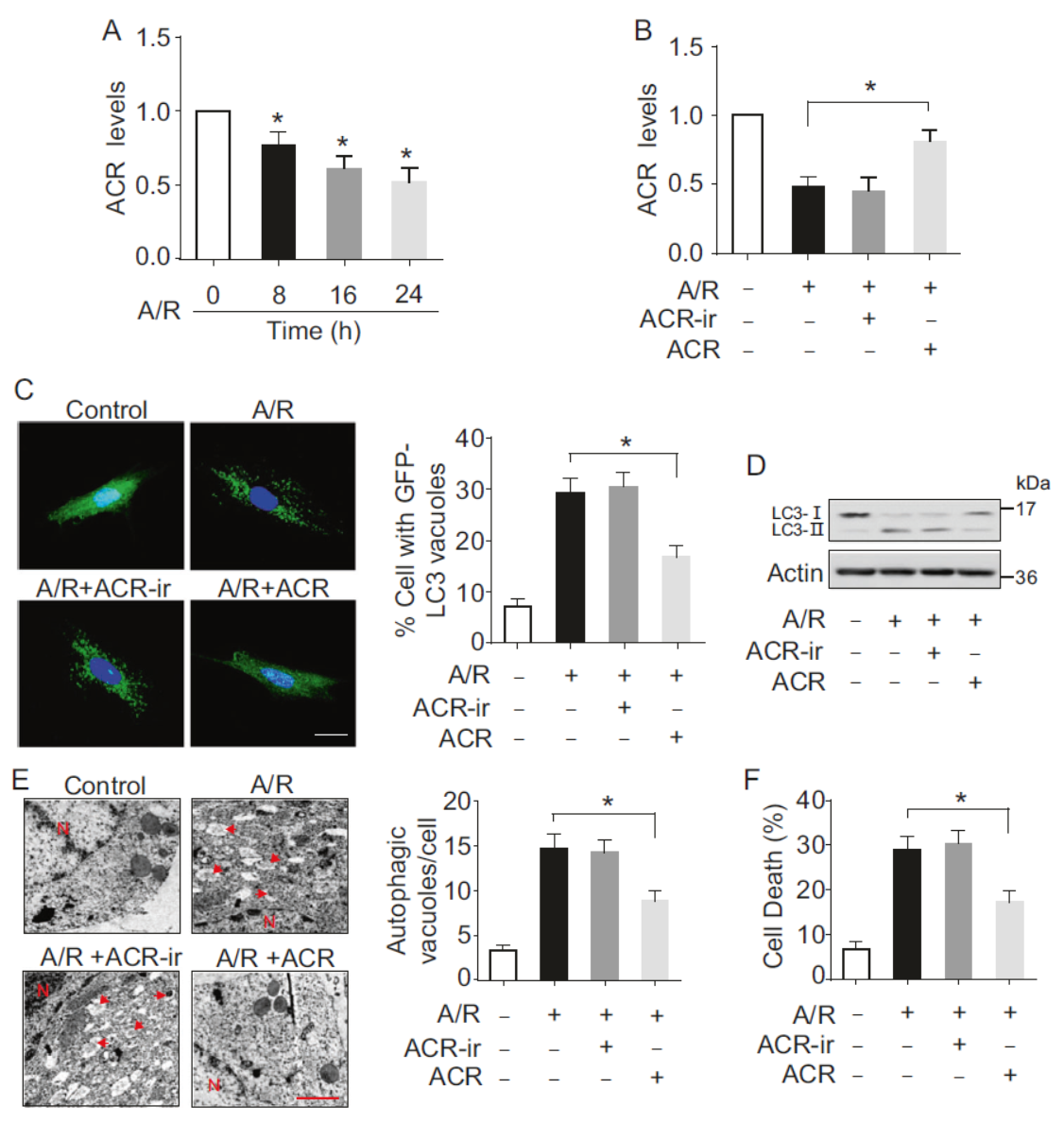
图2 过表达ACR抑制自噬 (引自[1])
敲低ACR后进行转录组芯片分析,探索与ACR相关的下游通路分子。结果表明在敲降ACR后,Pink1和Irgm1显著下降,QPCR和Western也验证了这一变化。但过表达ACR后Pink1增加的非常明显,Irgm1变化不显著。因此将ACR相关的通路锁定在对Pink1的调控作用上。
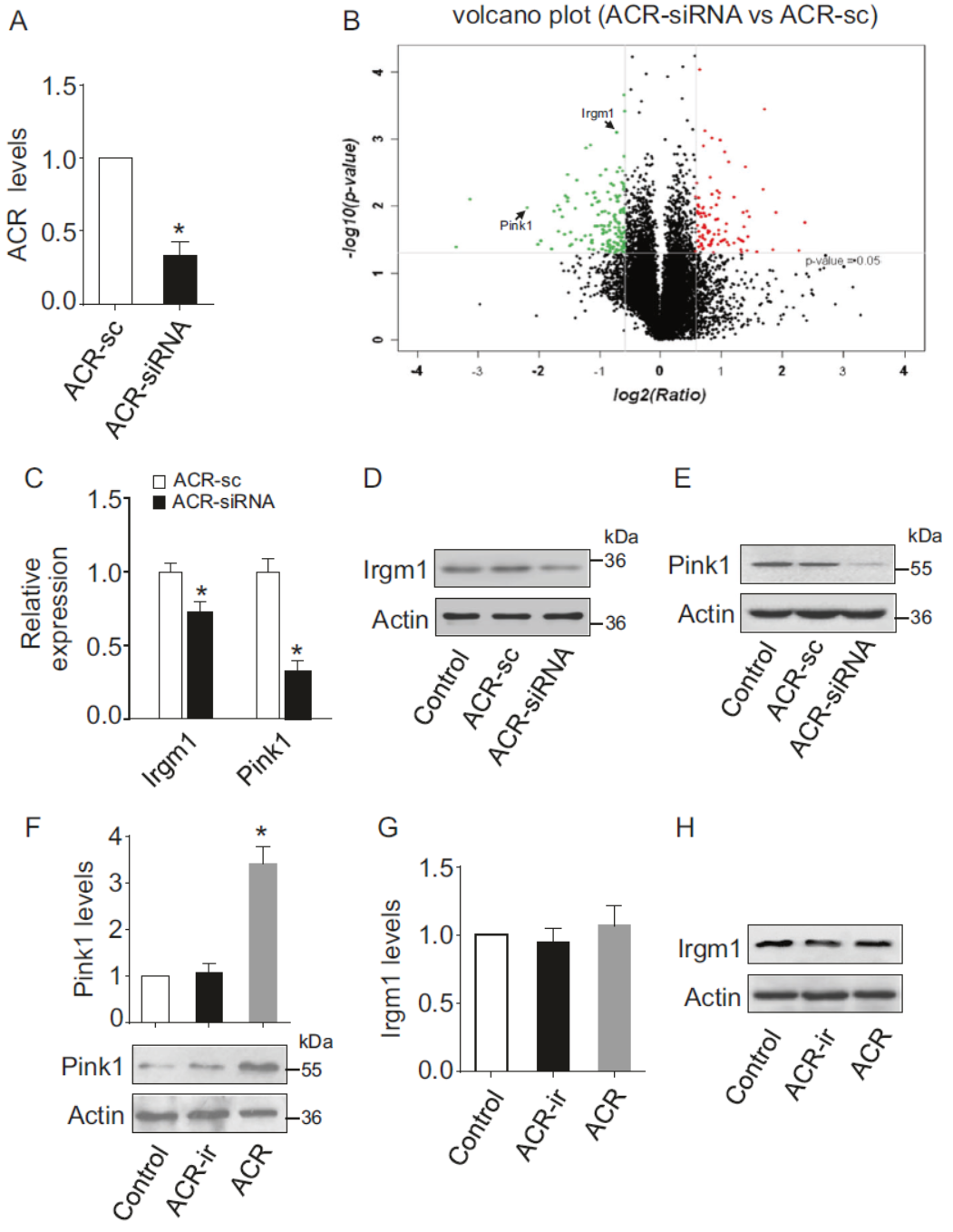
图3 ACR调控分子分析 (引自[1])
作者发现Pink1的启动子存在CpG甲基化区域,因此怀疑Pink1的表达是通过启动子甲基化状态调控的。DNA甲基化抑制剂5-Aza处理后能显著增加Pink1的表达,进一步佐证了这个机制。通过抗Dnmt1,Dnmt3a,Dnmt3b的抗体分别RIP实验,表明只有Dnmt3b能够富集ACR。靶向ACR的探针Pull-down也能够特异性富集Dnmt3b。敲降ACR后Pull-down实验中Dnmt3b的富集减弱。Dnmt3b是否可以靶向Pink1的启动子并调控其表达呢?通过ChIP实验证明,Dnmt3b可以靶向结合Pink1的启动子,敲降ACR后结合变强。干扰Dnmt3b能够增加Pink1蛋白水平。A/R和I/R处理后靶向Pink1的Dnmt3b显著增多。这些实验表明ACR通过结合Dnmt3b,抑制其与Pink1启动子的结合。
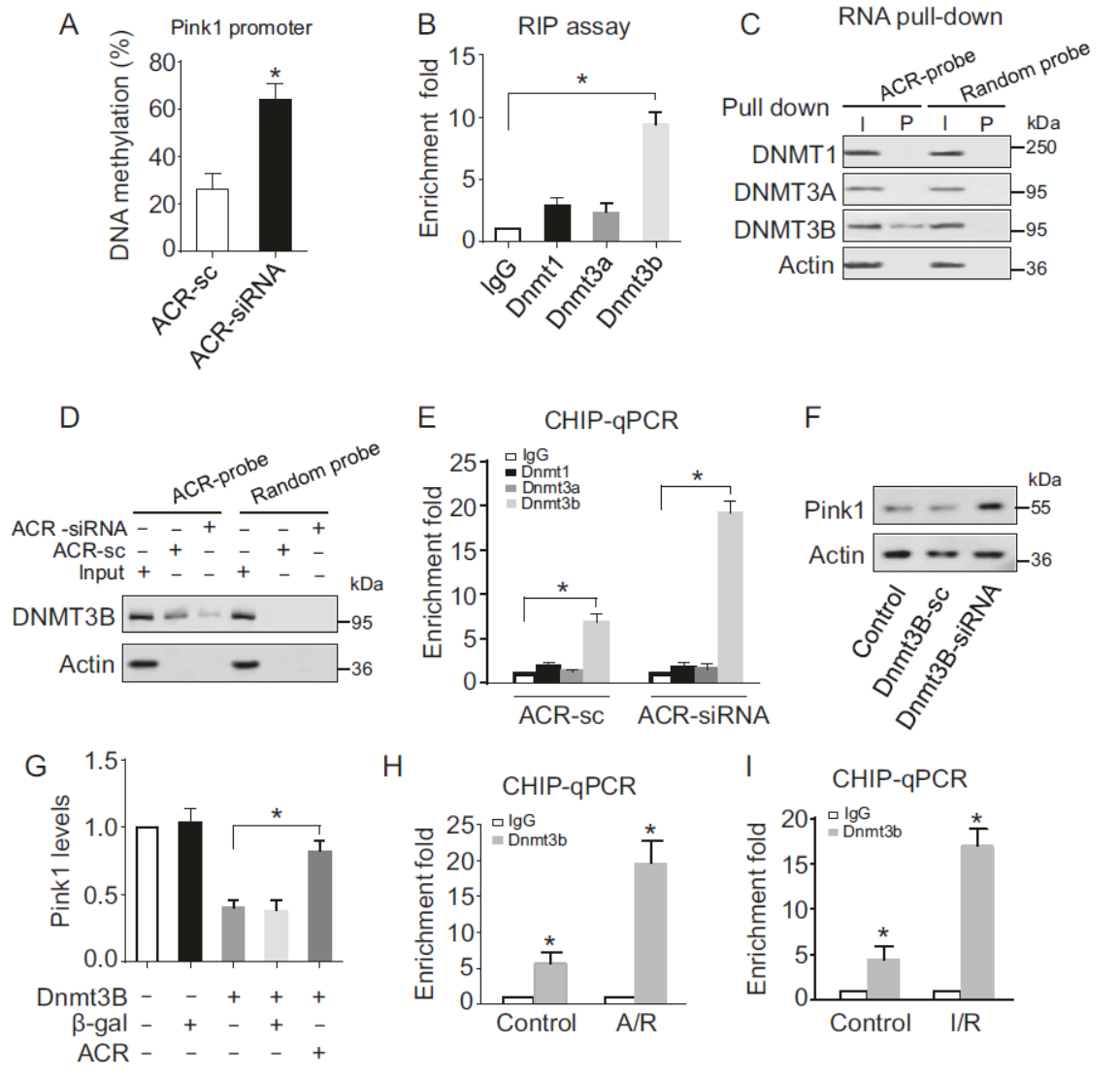
图4 ACR通过Dnmt3b调控Pink1启动子甲基化状态 (引自[1])
进一步,作者在体外和体内证明了Pink1调控自噬通路。为了找到Pink1调控自噬过程的机制,作者利用CoIP后Western鉴定的方法证明Pink1与FAM65B相互作用。基于质谱分析和抗体验证,证明S46是Pink1靶向FAM65B的位点
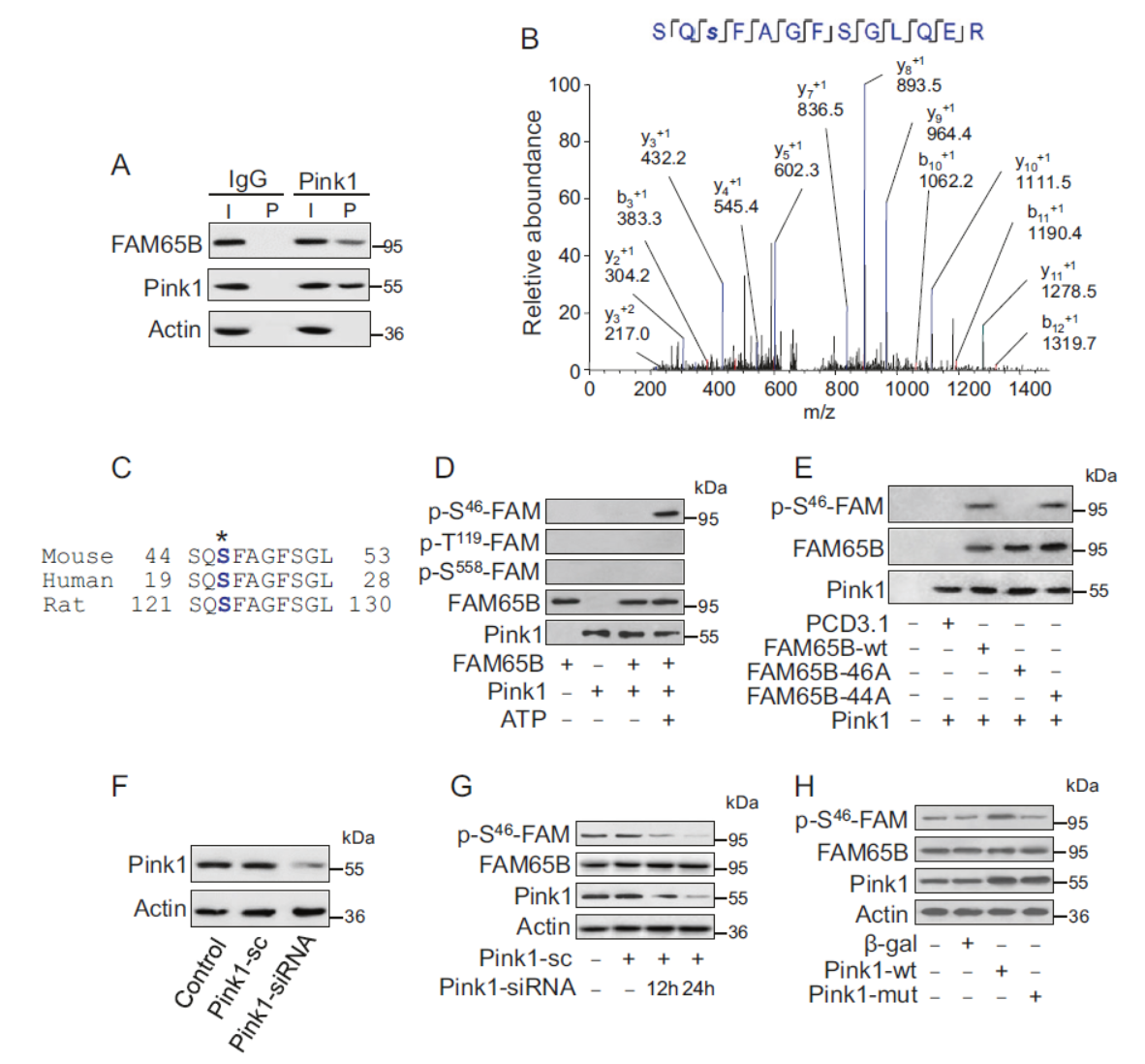
图5 Pink1靶向调控FAM65B的磷酸化 (引自[1])
本文的工作比较细致严谨,环环相扣,一步步证明了ACR与Dnmt3b相互作用,抑制后者结合Pink1的启动子。在缺氧/复氧或心肌缺血再灌注条件下维持细胞内Pink1的水平,抑制自噬通路的过度激活。本文的机制属于竞争性结合蛋白的“protein sponge”模型,值得广大同行学习借鉴。
circAGO2参与RNA稳定性调控
AGO2是RISC复合物中唯一具有内切核酸酶活性的亚基成分,发现在多种肿瘤中呈现异常高表达的状态。在这篇文章中作者发现了来自AGO2内含子的一种circRNA(circAGO2)能够结合HuR蛋白,促进HuR在靶基因的3’UTR富集,阻止AGO2的结合,调控靶基因的稳定性。
circBase数据库中AGO2基因来源的circRNA共有6种,在胃癌AGS中能检测到其中的两种,其中一种(hsa_circ_0135889)在前列腺癌和结肠癌和胃癌组织中明显高于癌旁组织,将其命名为circAGO2。Sanger测序鉴定其序列,FISH分析亚细胞定位表明circAGO2主要定位于细胞质中。常见的细胞系中进行Northern实验表明在肿瘤细胞中存在明显的circAGO2高表达,其分子量也与预期一致。
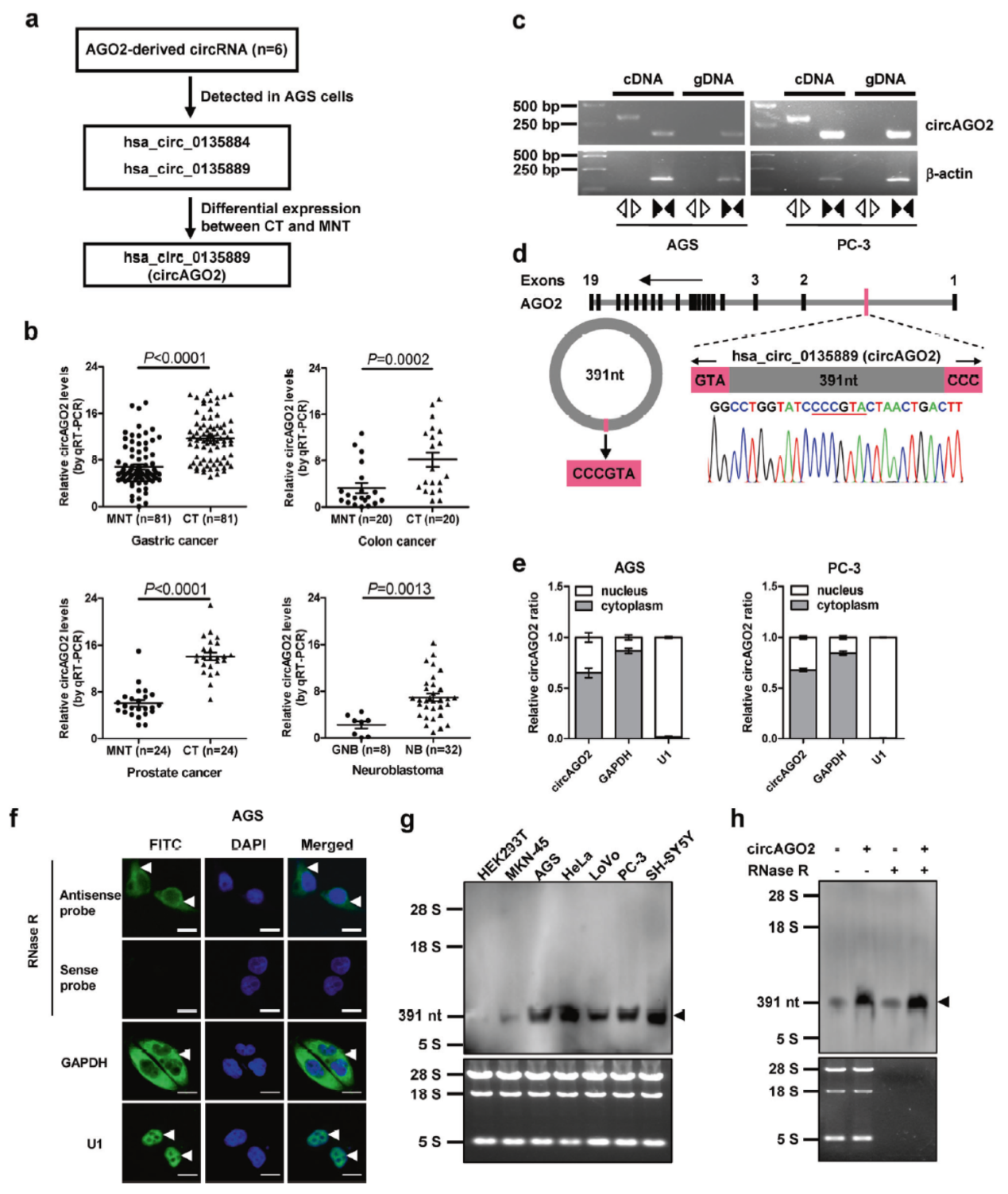
图6 circAGO2鉴定 (引自[2])
MKN-5细胞中过表达及AGS中干扰circAGO2表明circAGO2的表达与细胞侵袭明显相关。皮下成瘤实验也表明高表达circAGO2后MKN-5成瘤增强,干扰circAGO2后AGS细胞成瘤减弱。尾静脉注射两种细胞也证明circAGO2表达与肿瘤迁移密切有关。
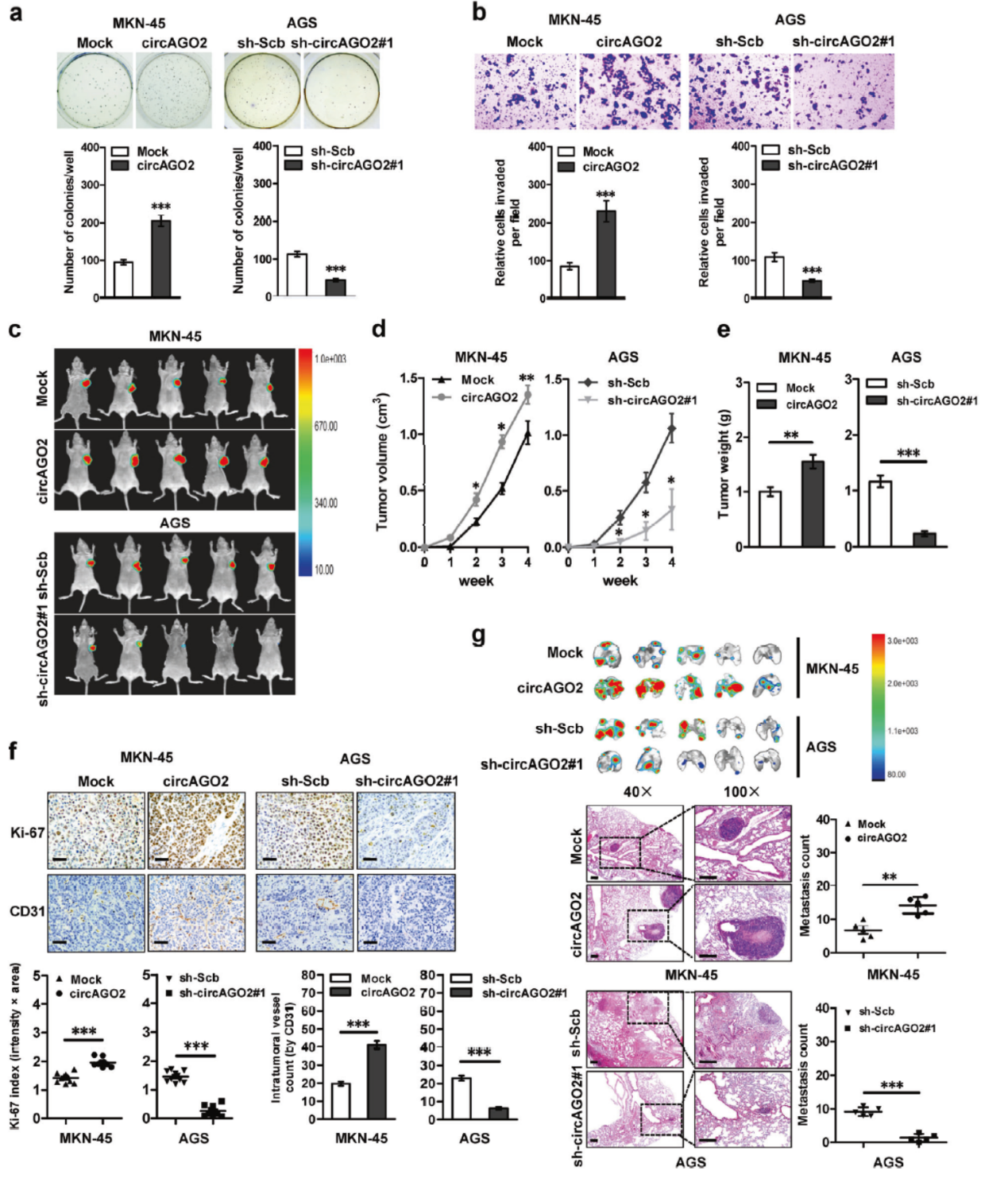
图7 circAGO2促进肿瘤迁移 (引自[2])
Pull-down实验分析与circAGO2互作的蛋白分子,质谱分析找到767种circAGO2互作的蛋白分子,结合数据库检索RBP蛋白及已知的AGO2结合蛋白,锁定了四种潜在的互作蛋白:TARDBP, RPL27A,RBM4和HuR。但进一步的Pull-down验证表明只有HuR能够特异性的在circAGO2探针捕获分子中富集。电泳迁移率改变分析(EMSA),GST融合标签Pull-down, FISH共定位分析也证明了circAGO2与HuR相互作用。
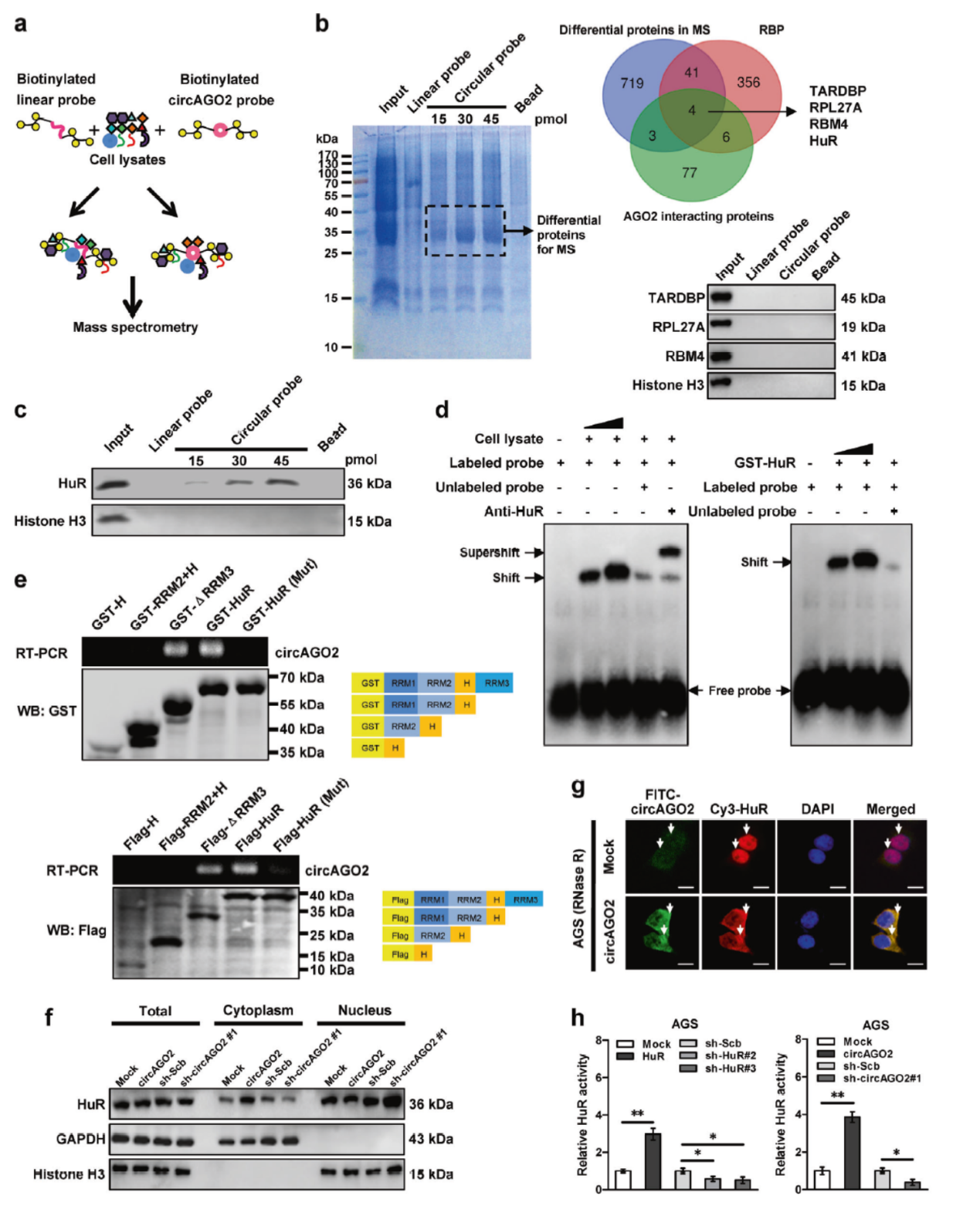
图8 circAGO2与HuR相互作用分析 (引自[2])
CoIP实验证明HuR与AGO2有相互作用。过表达和干扰circAGO2后转录组分析结合CLIPdb数据库的信息,表明有132种基因3’UTR存在AU-rich的序列(ARE)。miRWalk工具分析了这些ARE附近的结合miRNA,RIP分析表明HuR与AGO2均可与这些基因的3’UTR结合,过表达circAGO2促进HuR与靶基因的结合,但同时会抑制AGO2的结合。结合干扰HuR,过表达miRNAs。在多种细胞中验证了这一现象。
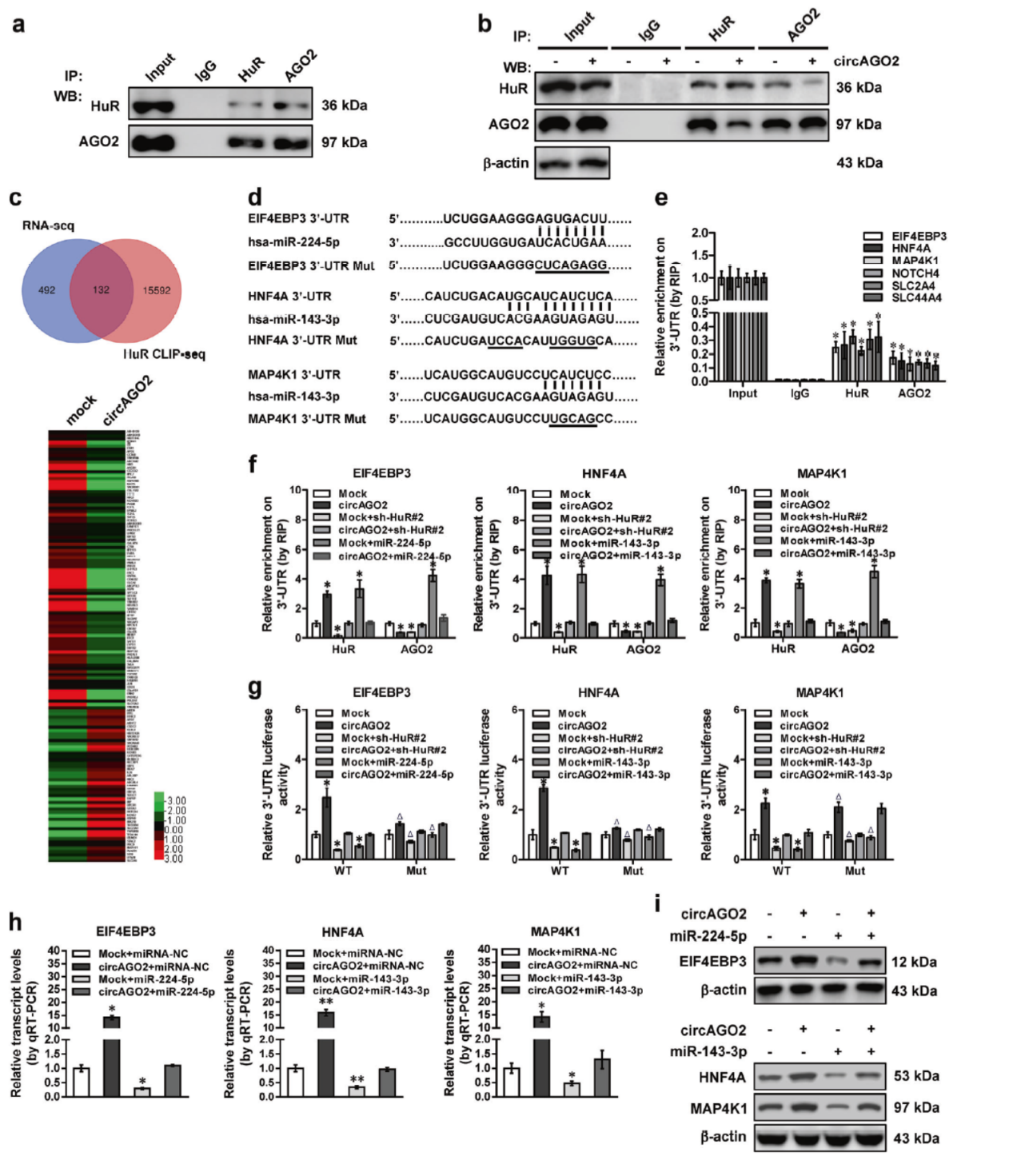
图9 circAGO2调控HuR与AGO2的功能 (引自[2])
作者还进一步分析了稳定干扰circAGO2对成瘤能力的影响。基于HuR与circAGO2相互作用的分子模拟,设计了可阻断该相互作用的小肽HIP-13,实验表明该多肽能有效竞争性结合内源circAGO2,阻碍其与HuR的结合并抑制肿瘤迁移作用。
本文的故事丰富了我们对AGO2调控靶基因的机制的认识,也证明了circRNA是一类重要的功能分子,对探索circRNA的机制和功能提供了很有价值的借鉴。
参考文献:
1. Lu-Yu Zhou, M.Z., Yan Huang, Sheng Xu, Tao An, Yun-Hong Wang, Rong-Cheng Zhang, Cui-Yun Liu, Yan-Han Dong, Man Wang, Li-Li Qian, Murugavel Ponnusamy, Yu-Hui Zhang, Jian Zhang, Kun Wang, The circular RNA ACR attenuates myocardial ischemia/reperfusion injury by suppressing autophagy via modulation of the Pink1/FAM65B pathway. Cell Death & Differentiation, 2018.
2. Yajun Chen, F.Y., Erhu Fang, Wenjing Xiao, Hong Mei, Huanhuan Li, Dan Li, Huajie Song, Jianqun Wang, Mei Hong, Xiaojing Wang, Kai Huang, Liduan Zheng, Qiangsong Tong, Circular RNA circAGO2 drives cancer progression through facilitating HuR-repressed functions of AGO2-miRNA complexes. Cell Death & Differentiation, 2018.
3. Huang, R., et al., Circular RNA HIPK2 regulates astrocyte activation via cooperation of autophagy and ER stress by targeting MIR124-2HG. Autophagy, 2017: p. 0.
4. Yang, L., et al., Engagement of circular RNA HECW2 in the nonautophagic role of ATG5 implicated in the endothelial-mesenchymal transition. Autophagy, 2017: p. 1-70.