文章:
Reconstruction of full-length circular RNAs enables isoform-level quantification
研究团队:
中国科学院北京生命科学研究院计算基因组学实验室
赵方庆团队
第一作者:博士研究生郑毅&助理研究员冀培丰
发表期刊:
Genome Medicine
发表时间:
2019年1月19日
引言
“君子生非异也,善假于物也。”这句话很适合该文的方法CIRI-full,它巧妙地应用了i)环状RNA的本性——环状且较短(大部分约300~500bp); ii)环化建库以及更长双末端测序(PE250, PE300)——环状RNA序列全覆盖。
名词解释
FRJ:forward splice junction,前向剪接位点,比如 mRNA 是pre-mRNA 通过剪接体复合物通过前向剪切去除内含子将外显子连在一起的产物。
BRJ:back splice junction,反向剪接位点,比如环状RNA通过剪接体复合物将供体3’末端外显子与受体外显子5’末端连接在一起的产物。
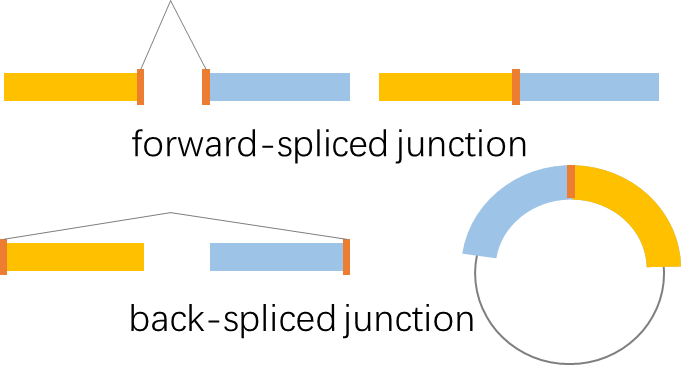
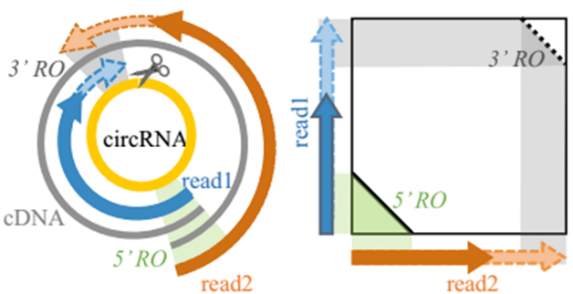
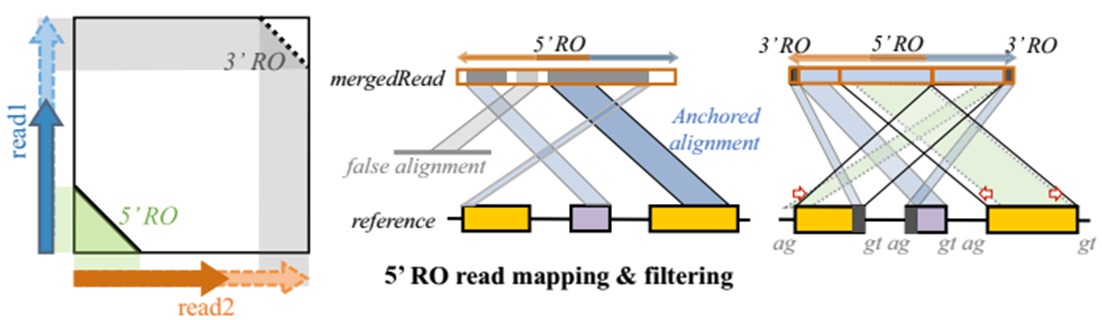
RO:reverse overlap,反向交叠,如图灰色阴影所示,表示双末端测序(paired-end sequencing) Read1 与 Read2 的5’末端或3’末端存在反向一致序列。
Cirexon:circRNA’s exon,构成环状RNA的外显子
Isoform:来自同一基因组区间的环状RNA转录本
FSG:forward splice graph,前向剪接图
文章亮点
- 提出了识别环状RNA全长序列的方法CIRI-full
- 相比基于BSJ的方法,RO方法对低表达丰度的环状RNA 更敏感
- 利用FSG的方法精确识别和量化环状RNA的转录本(isoform)
- 6个物种(包括人,恒河猴,小鼠,大鼠,兔,鸡)大脑全长环状RNA
- 相比BSJ,CIRI-full获得的转录本(isoform)能过滤假阳性的差异表达环状RNA
数据资源
测序数据:PRJNA475651
软件:https://sourceforge.net/projects/ciri
环状RNA研究的拦路虎——无法准确捕获全长序列
环状RNA 功能的重要性毋庸置疑,然而对其功能的探究仍举步维艰,主要原因除了人们仍局限于研究线性RNA分子的思维模式,另一个主要原因就是无法大规模获得环状RNA的全长序列并准确定量。
我们都知道,从序列同源以及物种进化地角度能够有效地探究一个遗传分子可能存在地功能;另外,一个遗传分子表达的准确定量是反应其在生物体内发挥功能的有效途径。然而,大规模从 RNA-seq 中识别全长 circRNA 的方法仍待开发。
这篇文章就提出了一个新的方法CIRI-full从RNA-seq中探究环状RNA转录本的全长序列以及表达定量。
实验基础
当插入片段长度(即测序文库片段大小)大于环状RNA序列长度,那么就可以观察到双末端 reads 的RO事件。
建库:环化建库,最后插入片段大小为300-800bp
测序平台:Illumina HiSeq 2500 platform
Read长度:250 x 2, 300 x 2
方法概述
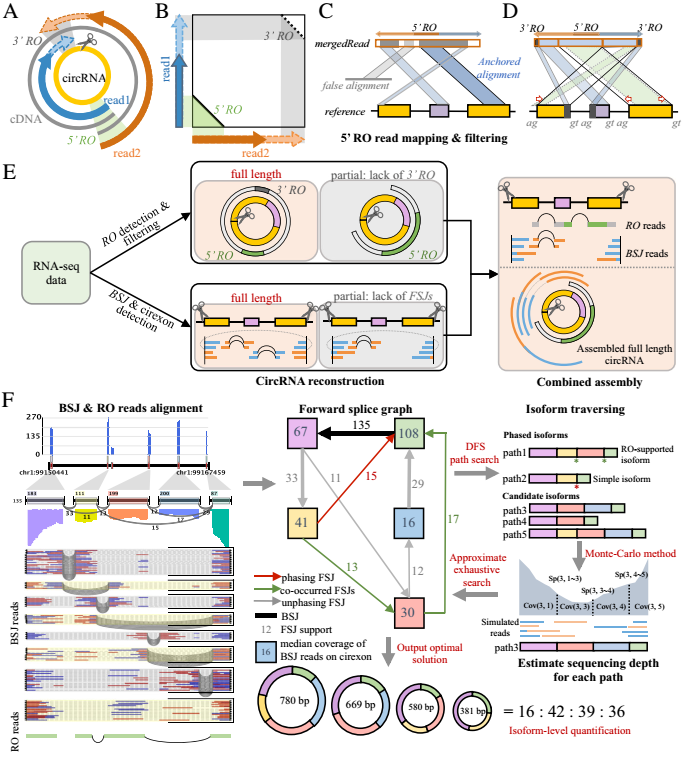
该方法总共分为四部分,包括
- RO read 的检测与检验(图A-D)
检测双末端 reads(paired-end reads)Read1与Read2的5’末端是否存在反向交叠RO(图A-B),如果存在则作为候选RO-merged read;随后分割比对RO-merged read 到参考基因组,最长的作为位置标签——锚定位点anchor,区分异常以及无法比对的区域(图C);校正剪切信号GT/AG确定环状RNA边界(图D)。
- BSJ 以及 cirexon 检测
利用 CIRI2 检测反向剪切事件BSJ,并通过CIRI-AS识别BSJ中的可变剪接事件(single-splice event)。如果构成BSJ的两个reads只落在BSJ内部的外显子即cirexons,那么环状RNA的全长序列就能由cirexons线性重构。
- 组装RO与BSJ的reads(图E)
- RO-merged read存在3’-RO或两个末端落在同一个cirexon上
- 每个BSJ的所有双末端reads都落在cirexons
以上两种情况可以直接确定环状RNA全长,而剩余的reads,CIRI-full结合两者优点——BSJ确定边界,RO-reads确定内部cirexons——进一步组装全长。
- 转录本isoform重构与定量(图F)
CIRI-full用BSJ cirexons以及RO-merged reads都构建了一个前向剪接图FSG,利用深度优先搜索算法(adapted depth-first search)将FSG分解成通道(path)——候选转录本(isoform),然后用蒙特卡洛仿真(Monte Carlo simulation)方法模拟每个通道(转录本)的表达丰度,最后用近乎穷举的方式寻找每一条通道相对表达丰度(设置了每个环状RNA最多10个转录本以提高效率)。
这里是分割线~
以下内容又臭又长!
方法详述
- RO read 的检测与检验
5’-RO识别策略 对于每对 Read1 与 Read2,Read1 与 Read2 其中一个的 5’ 末端的前 10 bp 被分成步长为 1 bp、窗口为 8 bp 的三份(如示例图,假设为 Read1 的 5’ 端)。

这些子序列被用来作为种子搜索 Read2 的 5’ 末端,一旦所有种子匹配碱基数 >= 7bp,那么从 Read2 的 5’ 最后一个碱基到 Read1 匹配到的那个位点的序列被提取出来,然后再比对到 Read1 上。如果满足以下条件,则这对 reads 被认为 5’-RO:reads pair上的长度 >= 13 bp,碱基一致性 >= 95%。随后,Read1 与 Read2 根据交叠部分合并为一条长 read 作为候选 RO-merged read以待进一步验证。3’-RO与此类似。
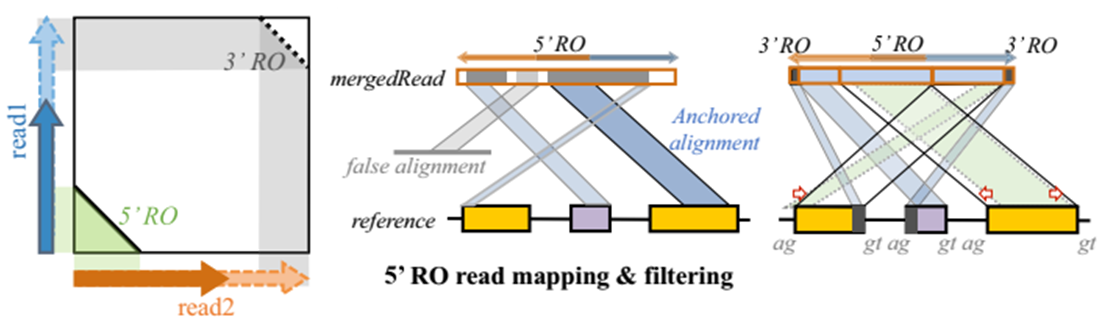
过滤假阳性 候选RO-merged read 通过BWA-MEM比对确定在参考基因组上的位置,然后将比对长度进行排序,其中最长且比对得分 > 15 的序列被作为anchor(锚定位点)来确定 reads 的位置。如果anchor 的两头在基因组上的范围 < 100Kbp,那么将计算RO-merged read 比对上基因组的长度;长度如若超过了 read 的一半,将用于再次做局部比对(local realignment)来确定RO-merged read在基因组上的精确位置——寻找BSJ(如果没有将用anchor),并用动态规划算法计算无法比对以及异常比对的片段。之后将过滤假阳性RO-merged read,原则包括:read不包含BSJ以及read两端等长子序列比对位置没有落在anchor 两侧。
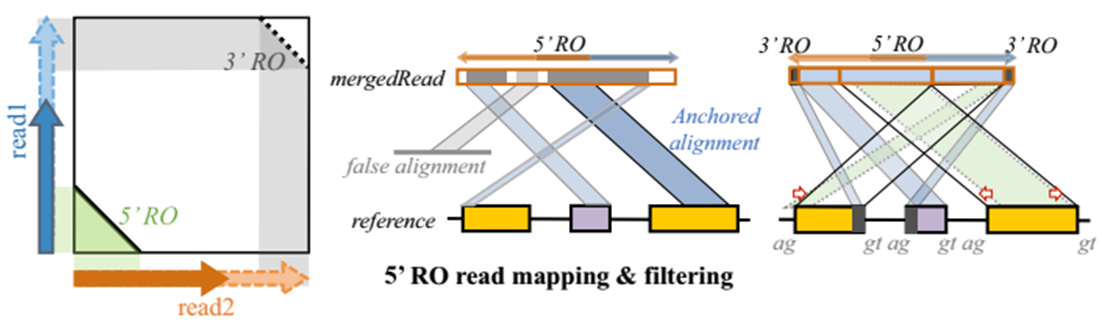
校正GT/AG 由于BWA-MEM无法获得GT/AG剪切信号以及精确的剪切边界,另外RO-merged reads可能包含套索结构(lariat structure)。因此,需要对每条候选RO- merged read检测和校正GT/AG位点,如果不存在将丢弃。另外,对每条read剪接位点(junction site)上下游5-bp比对质量进行检测,如果存在gap或错配,也将丢弃不用。
- BSJ 以及 cirexon 检测
BSJ 用CIRI2检测,而BSJ中的可变剪切事件(single-splice events)用CIRI-AS来推测。BSJ的检测方法在所有识别软件中都比较类似,简单描述就是截取read的5’与3’末端的子序列比对到参考基因组上,如果比对方向相反将作为候选circRNA。详细请访问CIRI 以及CIRI2原文。
每个BSJ中的cirexons通过CIRI-AS捕获的剪切事件推测。
- 组装RO与BSJ的reads
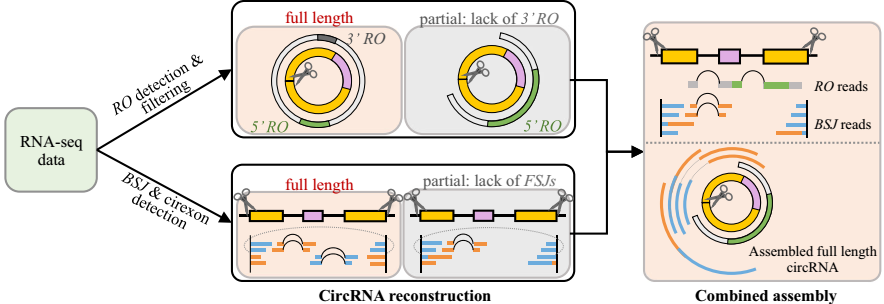 基于所识别到的RO-merged reads 以及BSJ边界信息,利用以下原则构建全长环状RNA。
基于所识别到的RO-merged reads 以及BSJ边界信息,利用以下原则构建全长环状RNA。
- 如果RO-merged read存在3’-RO或两个末端落在同一个cirexon上,那么该readd被认定为一个全长环状RNA;否则等待进一步组装。
- 如果每个BSJ的所有双末端reads都落在cirexons上,那么该BSJ的全长环状RNA由cirexons线性重构;否则BSJ以及cirexons等待进一步组装。
- RO与BSJ实际上存在互补信息——BSJ获得环状RNA精确的边界,而RO识别内部结构。
CIRI-full结合两者优点,将等待进一步组装的RO-merged reads以及BSJ reads根据BSJ进行排序聚类,如果两种read被发现落在同一个BSJ内,那么这些reads将被用来重构全长环状RNA。另外,RO-merged reads被用来确定BSJ未识别的额外cirexons,如果所有reads都落在cirexons上,那么全长将通过 cirexon 线性组装;如若不然,BSJ 所识别的 cirexons 将标记为部分重构的 circRNA。
- 转录本isoform重构与定量
对每一个全长环状RNA,CIRI-full用BSJ cirexons以及RO-merged reads都构建了一个前向剪接图FSG,其中节点表示cirexons,边表示两个外显子之间存在前向连接。
理论上,FSG包含了环状RNA所有存在的转录本,然而值得注意的是,由于环状RNA本身成环特性,所以FSG实际上是一个闭环。利用深度优先搜索算法(adapted depth-first search)将FSG分解成通道(path),迭代地从每个节点开始并在断点或起始节点(cirexon)结束;其中短通道被合并成更长的通道,而冗余的通道被过滤,为了避免过多可能的假阳性,CIRI-full设置了固定的最大通道数(默认10)。
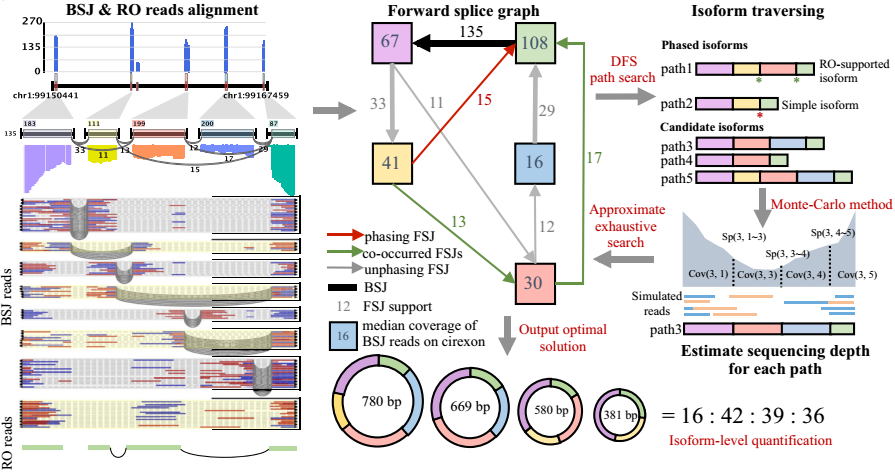
详细步骤:
- FSG的边包括4大类:
- BSJ
- Phasing FSJ(forward splice junction),表示只有一个环状转录本
- Co-occurred FSJs,表示剪切事件的数目与RO reads的数目相同
- 剩余的FSJ
其中,包含phasing FSJ和Co-occurred FSJs的通道优先级最高,被称为phased isoforms。而对第四类剩余的FSJs,根据节点的测序深度排序,只有top10被保留。所有保留的通道将作为候选转录本。
- 为了确定每条通道的相对丰度,用蒙特卡洛仿真(Monte Carlo simulation)方法根据 RNA-seq 插入片段的长度分布(通过 paired-end reads 在参考基因组上的距离推测)模拟每条通道的 BSJ-reads 分布
- 为了量化每条通道的相对丰度,CIRI-full 使用了穷举法(approximate exhaustive search algorithm):先为每条通道设置一个相对丰度(正整数),并且所有通道丰度和等于BSJ-reads总数。基于初始相对丰度以及模拟BSJ-reads,每条通道节点与边的累积丰度被计算;根据mapped BSJ-reads计算节点与边的真实丰度;随后,计算模拟值与真实值的距离。随后迭代地跟新模拟值,知道距离趋于收敛。
通过以上步骤,最终获得每个环状转录本的相对丰度。