具有完整闭环结构的环状RNA(circRNA)最初在1976年被鉴定。然而,由于传统RNA检测方法的限制,这些没有poly-A尾的RNA分子长期被忽略。近年来,随着高通量测序技术的发展,在真核转录组中发现了大量的circRNA 分子。由于circRNA具备高度组织表达特异性和跨物种保守的特点,目前已成为RNA分子家族中一颗耀眼的明星,目前越来越多的研究证据表明circRNAs在人类癌症和其他各类疾病中发挥重要作用。
结合circRNA高通量测序数据和现有的相关数据库可以对实验模型中circRNA分子网络进行数据深度挖掘,为后续实验验证提供可靠的实验依据和新思路,本文主要从ceRNA分子网络理论出发,解析circRNA的一般数据分析流程和研究思路。
竞争性内源性RNA(ceRNA)一般指的是可海绵样吸附miRNA的转录物如mRNA、lncRNA、circRNA等分子,这些RNA分子通过与共享miRNA的竞争性结合在转录后水平上相互调节。最近,circRNA被证明具有丰富的保守miRNA反应元件(MREs),已成为ceRNA家族的新热点。关于circRNA吸附miRNA研究报道已占circRNAs发表文献的半壁江山。如circRNA经典分子ciRs7,生物信息预测发现ciRs7上存在非常多的miR-7结合位点,后续实验也证明环状RNA ciRs7竞争性吸附miR-7后,释放了miR-7下游一系列靶基因,在多种人类癌症中发挥重要作用。但circRNA数量众多(circbank数据库目前已收录人类circRNAs达到14万多种),还有许多未知circRNA仍需探索。
CircRNA分子的ceRNA网络分析主要从以下几个方面进行:
1、获取实验模型相关的RNA分子表达谱
CircRNA表达谱的数据可以从多个来源获取,除了自己对实验样本进行高通量测序或芯片检测外,还可以通过GEO,TCGA等公共数据库下载已有研究的高通量数据。在收集好需要的circRNA高通量原始数据后,最主要的目的就是获得实验分组间差异表达的circRNA分子,这里应用最广泛的就是R语言中的各种基因差异表达分析软件包,如edgeR和DESeq2两个软件最为主流。展示circRNA分子表达情况的统计分析图主要有热图、火山图和circos圈图等。
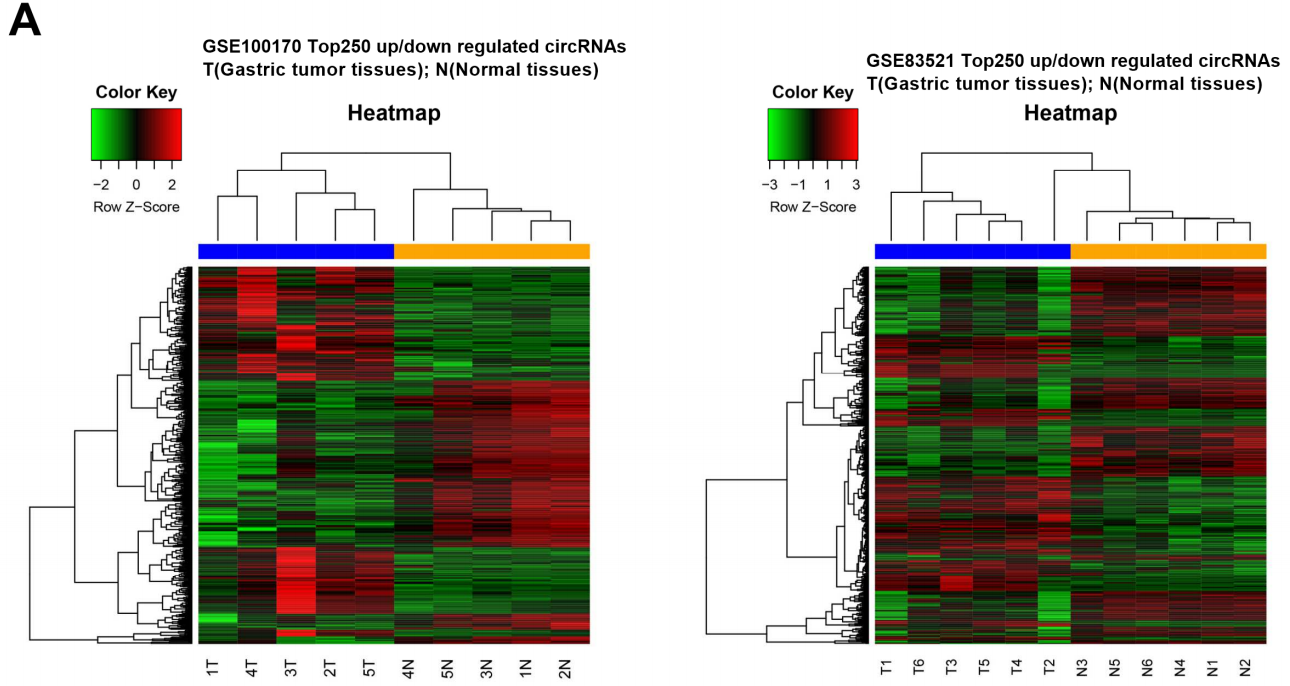
图注:胃癌中circRNA表达谱热图(基于GEO数据分析)PMID: 30419346。
PS:
GEO数据库:GEO(Gene Expression Omnibus),即基因表达数据库,美国国立卫生研究院NCBI于2000年创建的公共数据库,具有强大的灵活性和开放性,用户可以提交、储存、检索和下载多种形式的数据。GEO数据库是目前最大、最全面的公共基因表达数据资源。
TCGA数据库:肿瘤基因组图谱(TCGA)计划是由美国National Cancer Institute(NCI)和National Human Genome Research Institute(NHGRI)于2006年联合启动的项目,研究的癌症类型从最开始的多形性成胶质细胞瘤(GBM)到现在为止共有39种,涉及29种癌症器官,1万多个肿瘤样本,27万多份文件。
LncRNA,mRNA和miRNA分子表达谱的获取,同理,也可以通过实验或挖掘数据库获得。目前去核糖体的转录组测序,构建一次文库测序后即可分析得到circRNA,lncRNA和mRNA的表达谱,从时间和经济上来说非常划算,如果想得到miRNA的表达谱,只需要用同一份样本进行miRNA测序即可,这样就可以通过生物信息学分析构建ceRNA调控网络。当然如果手头经费有限,可以考虑利用GEO或TCGA这样的高通量数据库进行数据重分析,挖掘有价值的新分子或通路。如果研究模型是肿瘤,TCGA数据库绝对是数据挖掘的绝佳对象,TCGA对29种癌症进行了几乎全大规模转录组测序,包括lncRNA,miRNA和mRNA表达谱,还有基因甲基化测序相关的数据,样本多达1万多个,绝对是肿瘤分子生物学研究的金矿。
2、miRNA结合位点预测
ceRNA的核心理论就是基于miRNA可以靶向结合mRNA,circRNA和lncRNA等RNA分子,因此miRNA结合位点的预测也是关键的步骤,miRNA靶基因结合位点预测目前已有很多算法和数据库,主要分为两类,一类是单纯算法预测,常用的算法有targetscan,miRanda和RNAhybird等,第二类是一些数据库收集了文献报道的有实验证据的miRNA靶基因关系(主要集中在miRNA靶向mRNA的数据),如Tarbase数据库等。上述两类方法也可以综合起来一起进行分析合并。
PS
miRanda
miRanda是最早的一个利用生物信息学对miRNA靶基因进行预测的软件, 由Enright等人于2003年设计开发. 作为最早的miRNA靶基因预测软件, miRanda对3′UTR的筛选依据主要是从序列匹配、miRNA与mRNA双链的热稳定性以及靶位点的保守性三个方面进行分析。
TargetScan
TargetScan和TargetScanS. TargetScan是Lewis等人[15]在2003年开发的一款用于预测哺乳动物miRNA靶基因的软件, 该软件将RNA间相互作用的热力学模型与序列比对分析相结合, 预测不同物种间保守的miRNA结合位点。
RNAhybrid
RNAhybrid是Rehmsmeier等 人在2004年开发的一种基于分析miRNA和靶基因间形成双链的二级结构, 从而预测miRNA靶基因的软件。RNAhybrid的算法禁止分子内、miRNA 分子间及靶基因间形成二聚体, 根据miRNA和靶基因间结合自由能探测最佳的靶位点。
Circbank数据库对约14万种circRNA进行了miRNA结合位点预测(运用了miRanda和RNAhybird两种算法),网站提供了友好的检索查询界面,针对每个circRNA以表格的形式列举出对应的miRNA分子。
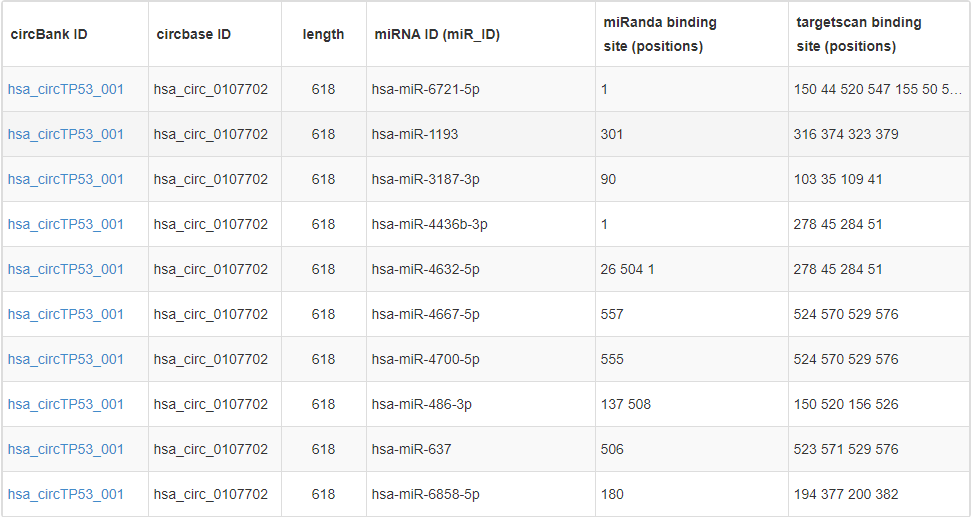
图注:circbank数据库预测hsa_circTP53_001可结合的miRNAs信息表
3、ceRNA网络构建
第3步就是构建circRNA,lncRNA,mRNA和miRNA分子网络,包括核心子网络提取和图形展示,这里主要用的到工具是cytoscape,cytoscape软件有丰富的插件,可以方便地从大型分子调控网络中找到核心节点子网络,图形展示上定制程度高,可以根据研究需要,将多种信息展现在网络上,如差异表达的分子用不同的颜色标记,连接线粗细表示关系强弱等。另外cytoscape中的一些插件,如BinGO可以进行gene ontology基因富集分析,CluePedia可以对pathway信号通路可视化操作等,cytoscape已经逐渐发展为分子网络的综合分析平台。
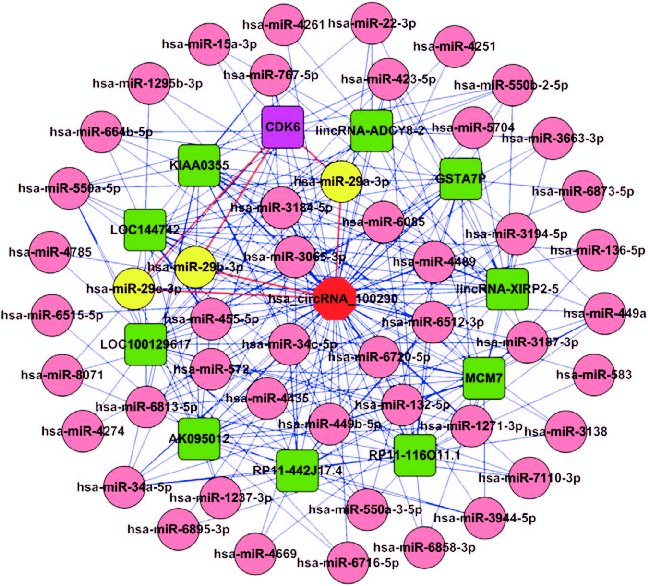
图注:circRNA_100290分子网络调控展示(PMID: 28368401)