2019年9月10日,bioRxiv平台在线了一篇关于将BLUEPRINT consortium基因表达数据分析用于成熟造血细胞,鉴定出细胞特异性新的lincRNA和circRNA的论文。本文的通讯作者为剑桥大学的Ernest Turro和Mattia Frontini教授。

简介
本研究使用来自脐带和成人外周血的27种成熟细胞的90个RNA样品以及来自8种成熟细胞的32个小RNA样品进行分析。通过使用贝叶斯差异表达分析方法来确定基因和转录本表达水平的变化,并鉴定细胞类型特异性的转录特征。作者通过使用RNA-seq的reads进行了引导转录组重建,鉴定了起源于400个基因间新基因的645个多外显子转录本,并且发现大多数新转录本具有低蛋白质编码潜力和高细胞类型特异性。此外,作者鉴定了55,187个circRNAs,也显示出非常高的细胞类型特异性,突出了非编码转录本在造血中的新作用。为了促进生物医学界对数据的探索和再利用,作者还提供了一个基于互联网的界面,允许绘制基因和转录本的表达模式,并下载标准化的表达数据(https://blueprint.haem.cam.ac.uk/bloodatlas/)。
研究内容
造血细胞转录组的复杂性
作者通过磁珠分离或流式细胞仪从72次全血和脐带血中分离了90个样品(表S1)。除了对血小板(PLT),嗜碱性粒细胞(BAS)和嗜酸性粒细胞(EOS)的去除核糖体RNA的RNA样品进行75 bp paired-end 测序以外,其余的样品平均产生了91 M的75 bp paired-end reads。每个小RNA样品平均产生了4.5 M 的50 bp single-end reads。长和短RNA的表达估计值对数的主成分分析(PCA)表明这些样品按细胞类型显示不同的聚类。
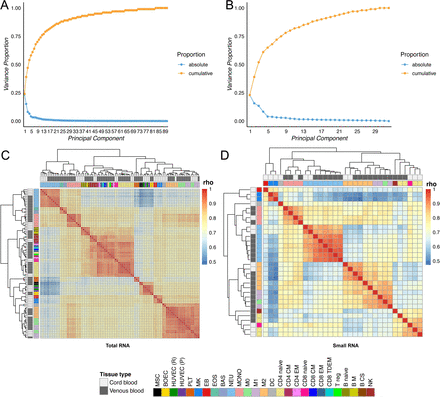
附图1 基因和miRNA的PCA分析和聚类分析
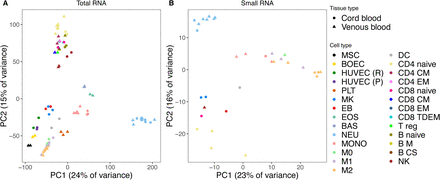
图1 基因和miRNA表达的主成分分析
就RNA分子的数量而言,一小部分表达的基因通常在任何给定组织或细胞类型的转录组中占主导地位。GTEx项目显示,全血被视为单一组织,其基因表达复杂性非常低,三个血红蛋白基因占总reads的60%以上。作者通过研究血液不同细胞类型中的转录组复杂性来完善此分析。由于线粒体基因在个体中的稳态表达差异很大,因此作者排除了线粒体基因进行基因表达研究。对于本研究中的除PLT外的所有细胞类型,占总表达量75%的基因的GO主要富集在一般生物学过程相关的功能类别,例如翻译或转录。然而,在PLT中,GO富集主要与止血,伤口愈合,凝血,血小板脱颗粒相关,而不是一般生物学过程。小RNA样品总体展示较低的复杂性。
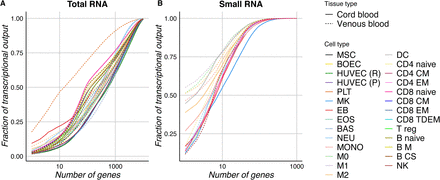
图2基因和miRNA转录组的复杂性
转录特征定义造血细胞功能
由于每种细胞类型中转录程度最高的基因均不编码该细胞类型的特定功能,因此作者认为这些功能一定由可能不高度表达但在整个造血过程中具有可变表达水平的基因编码。作者通过比较跨所有细胞类型的全局表达参数的统计模型与其中每种细胞类型的表达参数的统计模型来鉴定异源表达的基因。使用这种方法,作者发现了19,861个基因,占Ensembl中所有HGNC注释基因的59.5%,其差异表达的后验概率 > 0.8。然后差异表达基因根据表达量最大的细胞类型进行分类,即如果基因在表达量最高和第二表达最高的细胞类型之间的loge fold change > 0.1,则该基因则按最大表达量的细胞类型进行分类。
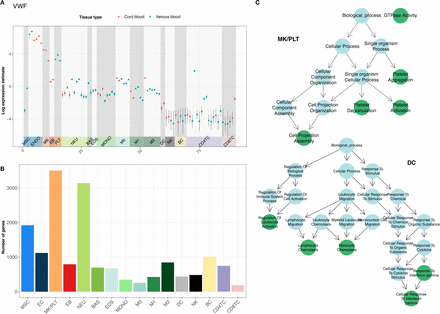
图3细胞类型特异性转录特征
miRNA的差异表达
作者将上述差异表达模型也应用于短RNA数据。结果,作者发现在2588个miRBASE注释的miRNA中有603个是差异表达的,后验概率> 0.8,其中有573个miRNA在表达量最高和第二表达最高的细胞类型之间的loge fold change > 0.1,因此被分类为特定细胞类型。使用现有的miRNA-mRNA相互作用数据库,作者并未发现miRNA的表达与其靶标表达之间的相关性,这与miRNA只是造血细胞转录调控中多种分子参与者中的一种有关。与其他研究结果一致,表明miRNA可以诱导mRNA翻译抑制而不会破坏mRNA。
转录组的从头组装鉴定新的基因和基因亚型
将本文测序的112个转录组数据从头组装,利用BLUEPRINT consortium中的转录组数据作参考,鉴定了起源于400个基因间新基因的645个多外显子转录本,其中,有368个的在至少一个样品中的log expression >0,这些基因可以根据细胞类型聚类,表明这些新基因可以作为鉴定细胞类型的标志;
通过 CPAT分数鉴定这些新基因大多数(545/645)是低编码能力,被鉴定为非编码RNA。由于ORF与转座子区域或重复序列或低复杂序列具有很少的重叠,所以作者根据这一特征,分析这些新基因中非编码(545/645)和具有潜在编码的序列中重复元件的分布,发现两者没有明显差异,表明在这些新基因中,即使具有较高的编码能力,它的特征也与非编码转录本的特征更接近,而不是与编码转录本的特征接近。因此,作者选择不将两组分开。
此外,新基因表达水平低于已知蛋白编码基因,并且与注释的lncRNA的表达水平相似。新基因还比注释的已知lncRNA和蛋白质编码基因具有更高的组织特异性。并且,新基因外显子序列的保守性较蛋白质编码基因差。这三个特性有助于解释它们的新颖性:新基因仅在非常有限的几种细胞类型中以低水平表达,尽管它们在生物学复制中始终保持一致。因此,仅在重建细胞类型特异性转录组时才使它们的鉴定成为可能。
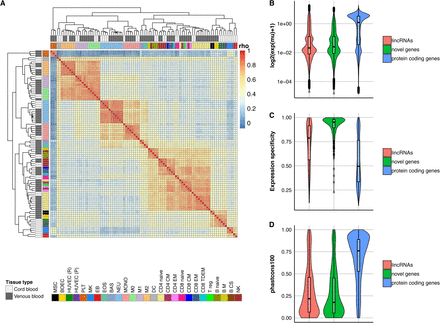
图4 鉴定的新基因的特征
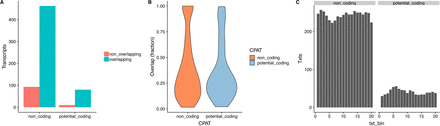
附图2 新转录本中重复元件的分布
成熟造血细胞中的circRNAs
作者通过CIRI, CIRCexplorer, find_circ, circRNA_finder和 PTESFinder 5种方法确定总RNA-seq数据中circRNA的丰度。要求通过至少三种方法来检测每个已鉴定的circRNA的反向剪接事件,以减轻序列比对软件的偏倚,并排除与基因组,多个基因或Ensembl 75注释的通读转录本中的已知片段重复重叠的预测。共鉴定了91,866个circRNAs,其中有55,187个在多个样本中检测到,且绝大多数反向剪接事件(81.64%)是外显子,并具有经典剪接位点。为了研究circRNAs在不同造血细胞中的表达模式,作者仅对在多个样品中观察到的circRNAs进行了成对相关分析和Spearman相关系数的层次聚类,表明circRNAs具有组织特异性。接下来,作者将circRNAs丰度与源自相同基因的线性RNA的表达进行了比较,并使用测度丰度比(AR),即每个位点的反向剪接读数计数比上规范连接数。
为了鉴定差异表达的circRNAs,作者对它们的丰度进行了成对比较,并鉴定了984个差异表达的circRNAs,并这些差异表达circRNAs的表达模式也按细胞类型进行聚类。尽管已经发现了非编码RNA的几种作用机制,但只有少数circRNAs被实验验证为功能性,其功能不同于其亲本基因,从而消除了GO分析的直接功能推论。
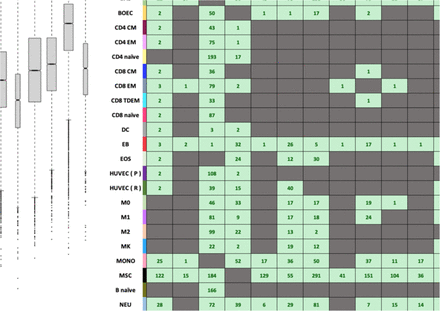
附图3 血细胞中差异表达circRNAs的丰度
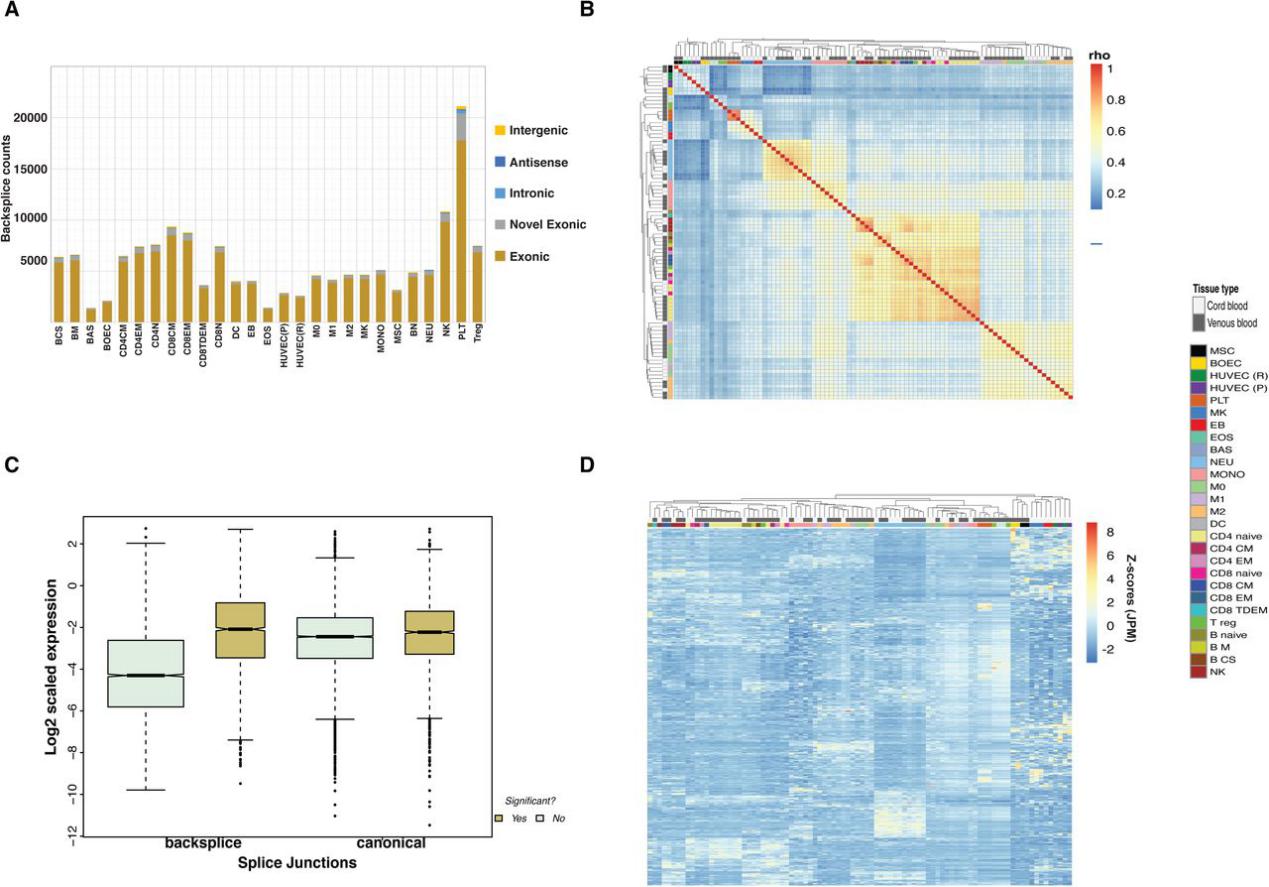
图5 CircRNA在血细胞中的表达
参考文献
Grassi, L., Izuogu, O. G., Jeorge, N. A., et al. (2019). Cell type specific novel lincRNAs and circRNAs in the BLUEPRINT haematopoietic transcriptomes atlas. bioRxiv, 764613. doi: https://doi.org/10.1101/764613