简介 环状RNA(circRNA) 是反向剪接和共价闭合的RNA。它们与RNA结合蛋白 (RBP) 的相互作用对许多疾病的发展有多重影响。目前已经有不少工具可用于识别circRNA上的RBP结合位点,但准确性、稳健性和解释性都不足。为此,中南大学计算机科学与工程学院王建新教授团队开发了一款全新的工具——CRMSS,一种基于多尺度特征序列和结构特征来预测 circRNA-RBP结合位点的方法。该项成果发表在了Briefings in Bioinformatics (IF=13.994) 期刊上。 算法优势 1. 不同的序列编码方法对性能的影响比较 CRMSS采用了k-mer算法,并与One-hot、CircRNA2vec、KNFP、Pseudo-coding四种序列编码方法进行了比较(表1)。结果显示CRMSS的k-mer方法获得了最佳性能。在其他编码方式下,预测性能都会有一定程度的下降,尤其是One-hot编码方法,导入数据的稀疏性导致性能出现明显下降。这些结果表明CRMSS的序列编码方法不仅更准确而且更稳定。 表1. 不同序列编码方案对基准数据集的影响 2. CRMSS与现有预测工具之间的比较 作者将CRMSS与目前最先进的四种工具CRIP、PASSION、CircSLNN和iCircRBPDHN在评估指标和统计分析方面,对基准数据集进行5倍的交叉验证。一方面,作者采用指定的评估指标来评估性能,测试了仅具有circRNA结构特征(RNA secondary structure, RSS)或仅具有RBP特征(RNA bindingDomain, RBD)的模型,以分析不同特征的影响,并检查了37个RBP上的所有结合位点。37个RBP的平均结果用于评估模型(图1)。可以看到,CRMSS的性能优于这四种工具,获得了最高的平均AUC值 0.899,并且在AUPR和Recall中表现得更好。 图1. 不同工具在37个RBP上的测试结果 另一方面,作者采用了Friedman检验和Nemenyi检验进行统计分析,并绘制了一个测试图,其中每个模型的平均排名值作为中点,CD 值作为线长(图2)。从图中可以看出,CRMSS的线与CRIP、Passion、CircSLNN的线没有重叠,这说明它们之间有很大的区别。与其他工具相比,CRMSS的线与iCircRBP-DHN的线接近,表明它们之间的差异较小。 图2. Friedman–Nemenyi检验对37个RBP的AUC测试结果 3. RSS和RBD模型对性能的影响比较 作者分析了RSS和RBD的影响,从图 1 中可以看到仅RSS或仅RBD会降低所有RBP的平均性能。然后,作者计算了AUC比率以获取RSS和RBD对每个RBP的影响(图3)。与RBD模型(Model 1)相比,RSS模型(Model 2)的性能下降更小,这表明RNA结构信息比RBP信息更能影响circRNA上的结合位点。 图3. RSS和RBD模型对每个RBP的影响 4. RBP结合性能的比较 为了进一步研究CRMSS模型是否捕捉到RBP的结合偏好。一般认为在相同的生物过程中,RBP往往具有相似的结合偏好。作者使用BLAST方法计算基准数据集中所有RBP对的序列相似性。图 4(A) 显示所有RBP对的平均序列相似性约为0.13。作者选择了两组序列相似性差异较大的RBP对进行对照实验,将具有最高序列相似性的10个RBP对设置为组1,而组2包含具有最低序列相似性的10个RBP对。从图 4(B) 来看,两组在序列相似性方面有显著差异。对于每个RBP对,作者用一个RBP的结合位点训练模型,然后预测另一个RBP的结合位点,预测精度反映了RBP对结合偏好的相似性。从图 4(C) 来看,组1中的RBP对的平均精度接近0.7,而组2中的RBP对的平均精度不超过0.3。因此,作者认为拥有更相似序列的RBP具有更接近的结合偏好。 图4. RBP结合性能的影响 5. 实际应用测试 为了测试CRMSS在实际应用中的表现,作者选取了两方面的数据集进行研究。一方面,作者从NPInter数据库中收集了经过实验验证的circRNA-RBP相互作用对。如图5所示,从所有的相互作用对中都检测到了结合位点,表明CRMSS的预测结果是可靠的。 图5. 预测实验验证的circRNA-RBP对上的结合位点 另一方面,作者采用RIP-seq数据集比较了CRMSS和上述四种工具的优劣(表2)。其中,被两种方法(Clirc和circRIP)检测到的114个结合位点被设置为可靠的结合位点,而两种方法都没有检测到的60个结合位点则设置为不可靠的结合位点。作者去除了577个结合位点的标签,将对应的circRNA和RBP信息输入到训练好的CRMSS模型中,并重新预测结合位点的标签。在114个可靠的结合位点中,约有107个可以被准确检测,CRMSS的准确率达到93.9%;在60个不可靠的结果中,只检测到两个预测的结合位点。从表2可知,CRMSS在所有工具中的表现最优异。 表2.不同工具在RIP-seq数据集中的检测能力 方法策略 对于circRNA,作者用k-mer编码circRNA序列,用RNAplfold计算circRNA的二级结构,然后将circRNA的两个特征连接起来。对于RBP,每个RBP都使用RBD的序列和结构频率进行编码。接着,作者分别将circRNA和RBP特征传输到由多尺度CNN层形成的残差块,输出被连接起来并传输到具有自主注意机制的BiLSTM。最后,作者使用softmax对具有完全连接层的结合位点进行分类。 图6.CRMSS方法概述 小结 综合上述所有的比较结果表明,CRMSS捕获到了结合偏好并给出了可靠的预测。与现有的RBP结合位点预测工具相比,CRMSS的性能表现得最优异,不仅提供了准确的预测,而且有助于进一步研究分子机制。感兴趣的研究者可以尝试下载使用。 参考文献Zhang L, Lu C, Zeng M, Li Y, Wang J. CRMSS: predicting circRNA-RBPbinding sites based on multi-scale characterizing sequence and structure features. Brief Bioinform. 2023 Jan 19;24(1):bbac530. 转载请联系邮箱授权:circRNA@163.com 分类: 最新重要进展admin二月 15, 2023评论

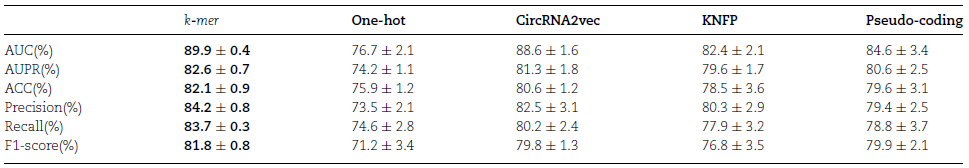
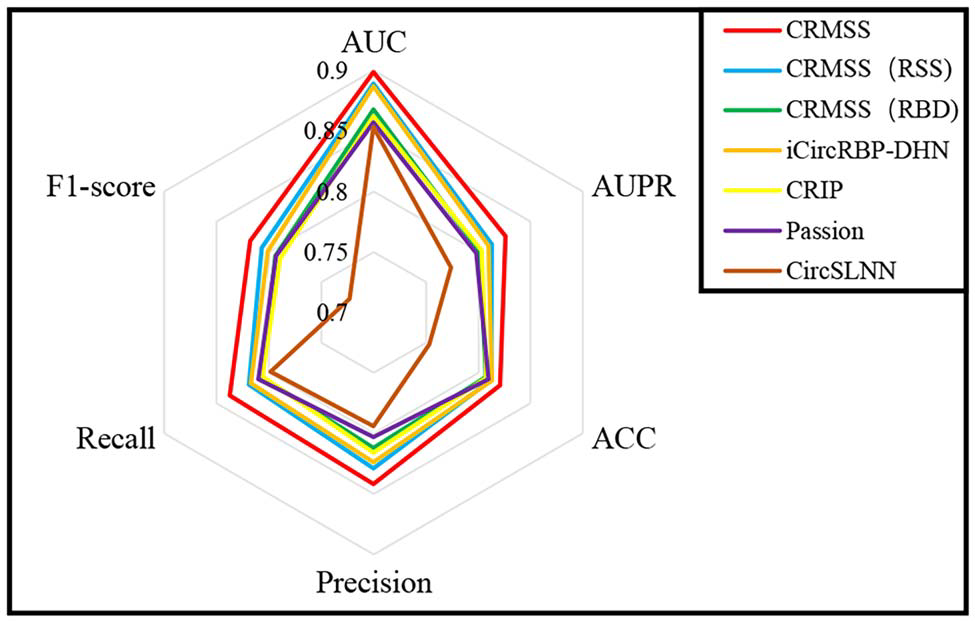
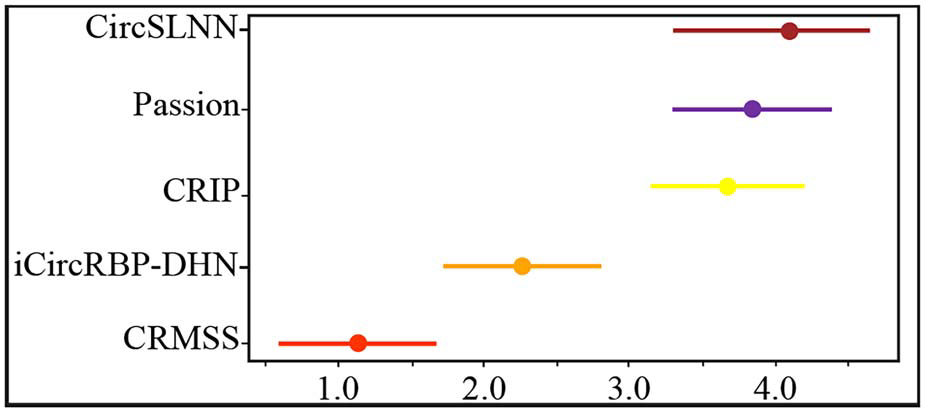
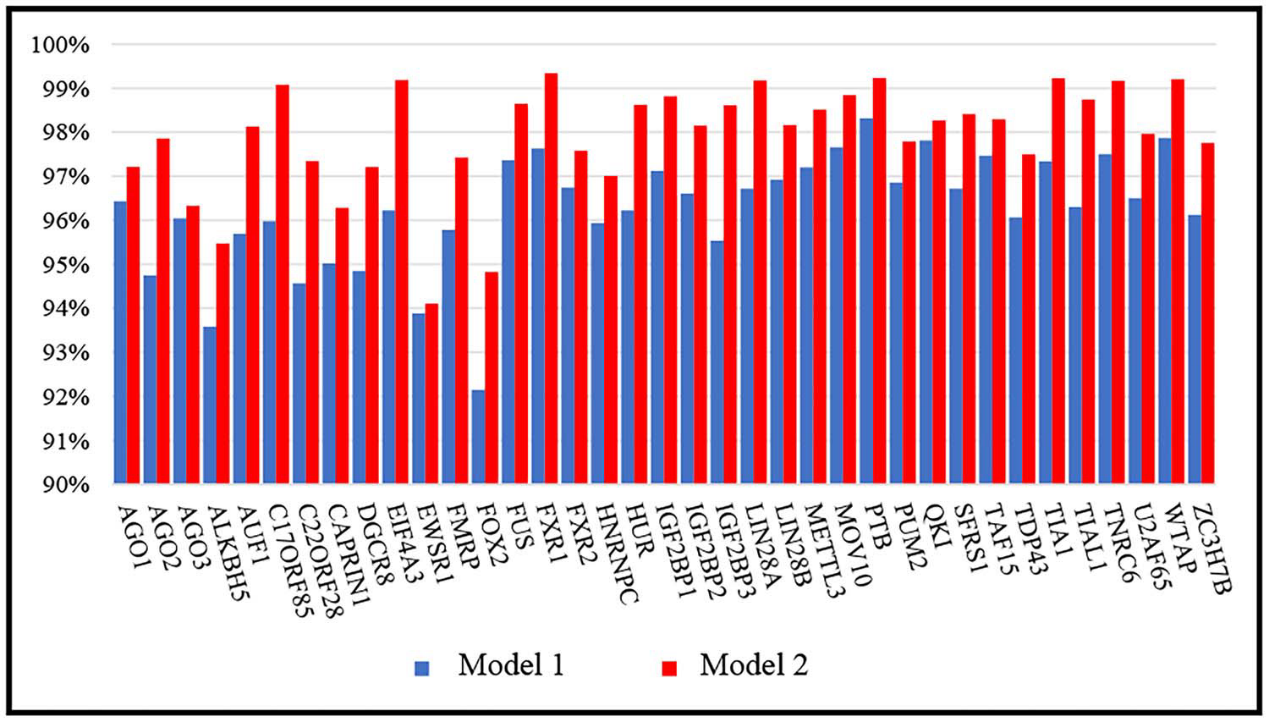


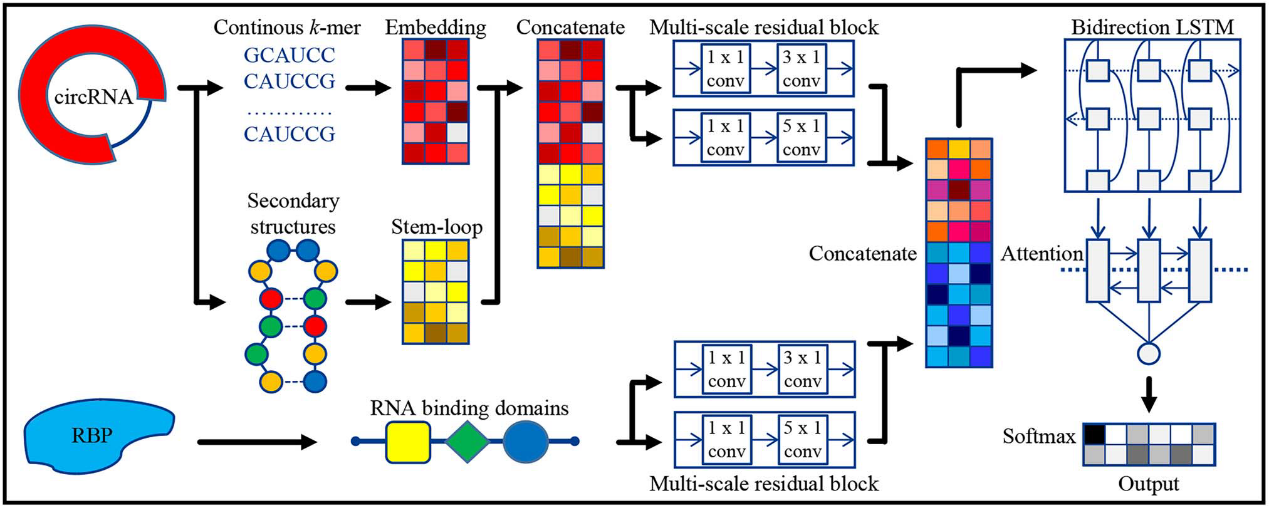 图6.CRMSS方法概述
图6.CRMSS方法概述